


HMGB 1 - One Protein to Rule Them All 1.5
A second part...kinda

This isn’t necessarily an extension of my HMGB1 article, or an updated comprehensive analysis on the One Ring of proteins, but it is both significant and impactful, with lasting consequences. I will preface this article as I lately do, I thought of writing a shorter article, but I think writing a longer one with the latest evidence in regards to HMGB1, and tying many aforementioned effects together.
You can read my first article below, which is paramount to understanding the following study. The article also has a significant amount of supplement recommendations on how to address HMGB1. I will provide a summary too.

HMGB1 is one of the most important proteins present in human cells, possessing an extensive list of primary, secondary, and tertiary functions, and it is a key mediator of inflammation. It is necessary for health, but as with everything, it is a problem in excess, highly sensitive to oxidative stress, and plays a role in many diseases and conditions. My focus in my article above was to cover the intersection between all the conditions in which SARS-CoV-2 plays a role and is implicated, and HMGB1’s role in them.
Like many of my favorite proteins, it possesses a dual-edge nature, and it often can seem paradoxical. Inside your cells, it will do specific, highly important functions, but when outside, it becomes a primary danger signal, alerting everything in its surroundings that death has come, as it is released in abundance when cells die. This is when it becomes a reactive, inflammatory (natural) chimera.
The general argument of my article was that it is not just that SARS-CoV-2 induces the expression and production of HMGB1 at a systemic level, but that HMGB1 interacts with the Spike Protein, and it was what enabled the Spike and virus to do a lot of the uncharacteristic, “novel” effects. One of the earliest names for HMGB1 was “Endotoxin-binding protein”, as initially believed to be its function. I hypothesized that HMGB1 binds to the Spike Protein, enabling it to form highly inflammatory, multi-faceted protein complexes.
A hypothesis no more.
Direct interaction of HMGB1 with SARS-CoV-2 facilitates its infection via RAGE-dependent endocytosis
Given the importance of HMGB1 in the progression and acceleration of so many diseases, its higher expression the more severe your infection is, the authors argued that HMGB1 is not just a marker for inflammation and severity, but potentially is an active facilitator of SARS-CoV-2. This is where something I called “Receptor Hierarchy” comes in, as the virus following biochemistry rules, it will primarily want to use the easiest and quickest way to infect your cells, ACE2.
It somehow can infect a myriad of different cells that do not express (meaning there isn’t much) ACE2 on their surface, thus the receptor hierarchy, using other receptors to infect cells, sometimes not using receptors at all. The authors themselves bring up the interaction between LPS-HMGB1, forming a potent inflammatory complex that interacts with RAGE, Receptor for Advanced-Glycation End-products, a receptor that can interact with a range of proteins, fats, and especially glycated ones, meaning when sugar is added to molecules.
Whenever small sugar molecules (glycation) are added to other proteins or lipids, it changes how they act/react. There are other forms of -ations, the process of addition of different “things” to active molecules, but glycation in the context of viruses, and long-term health are the most important, as we live in a carbohydrate abundance in our modern diets. Glycation can shield viruses from your immune system, and this can also impact how the body “eats and dissolves” the dead and debris of pathogens.
The goal of the researchers from the start here was to determine if HMGB1 could bind with SARS-COV-2 Spike Protein. They used Surface Plasmon Resonance (SPR), which is a way to measure molecular interaction in real time. HMGB1 consists of 3 parts, Box A which acts as an HMGB1 antagonist, Box B, which is the inflammatory part binding to TLR4 and RAGE for example, and C-Tail giving structure to the protein, and when fused with Box A it enhances its effects, C tail also plays a role in DNA binding, which both A and B do, influnencing how HMGB1 can bend DNA.
They use the entire protein and each part, not only trying to see if HMGB1 binds to the Spike Protein, but also which part actually does it. The full-length (entire) protein had a solid, strong binding score (the lower the score, the stronger it is), Box B had a significantly stronger value, the C Tail had almost as strong as Box B, with Box A having a much weaker interaction.
Next was testing if HMGB1 bound specifically to the S1 part of the Spike Protein, which arguably could be considered the “most toxic” part of the protein. C-Tail and the full-length protein both contained the RAGE-binding site. After observing the interaction, their next step was to assess how this affects SARS-CoV-2 infectivity.

They used a cell line that expresses very little ACE2, and also excluded the possibility of HMGB1 binding to ACE2. The more HMGB1 added to their test, the more Nucleocapsid Protein and higher viral load was observed. They also pre-incubated the virus with HMGB1 with a cell line that has almost no ACE2, with these cells exhibiting much higher levels of the virus, indicating increased infectivity in a HMGB1-SARS-CoV-2 complex manner.
Proceeding with their testing, they tested how each part of HMGB1 affects this higher infectivity, and as the full-length protein and the C-Tail enhanced infectivity, both parts containing RAGE-binding sites. Next, they aimed to assess the mechanism by the SARS-CoV-2- HMGB1 complexes enhance infectivity, focusing specifically on RAGE, using cell lines with high levels of RAGE and removing any expression of ACE2.
Incubation of HMGB1 with SARS-CoV-2 drastically increased the virus infectivity, without any ACE2. By using a form of decoy (soluble RAGE), they observed a reduction in infectivity, and the same observation was seen by using Azeliragon, a drug that lowers RAGE levels.
When extracellular HMGB1 forms a complex with Endotoxin (LPS), this jointed protein can bind to RAGE, which is expressed on the cell surface, enabling endocytosis. Endocytosis is a process by which a protein, or in this case a virus, can enter a cell or hijack it, usually bypassing “normal steps”. This is what the authors observe in their last step. Worth noting that in one of their tests, they used Chloroquine, a known inhibitor of endocytosis. Chloroquine could be called the parent molecule for the infamous drug HCQ, Hydroxychloroquine.
They observe these effects in vivo, using mice.
The One Ring to Rule Them All
Every few months, a groundbreaking paper regarding SARS-CoV-2 is published, usually to little noise, little social media virality (unless talking about mRNA vaccines), but providing groundbreaking data or evidence. The focus of the authors of this seminal paper was infectivity, if and how SARS-CoV-2 could subvert a ubiquitous protein in our bodies to infect more cells, and increase its replication threshold.
Research has to be done in steps, but to me, similar to both the paper providing evidence that SARS-CoV-2 Spike Protein acts as an Endotoxin Delivery system, and the paper providing evidence of the Spike Protein's superantigenic behavior, this is one that stands with them, and adds to the first.
If you read the primary article on HMGB1, you could go on a little exercise of your curiosity and find any condition HMGB1 plays a role and add SARS-CoV-2 to it, and you may be perplexed by what you find. HMGB1 will play a role by hook or by crook in a myriad of effects this chimera induces. This interaction is especially important with a few of the most important, lingering open questions in regards to this virus.
While SARS-CoV-2 can use as many receptors as I can remember, it possesses some lasting and unique effects in the human body, either in your veins, your brain, organs, but most importantly, our bone marrow. It is one of the most important mediators in the bone marrow, it can enhance immune suppression via complex interactions. This can explain why there is a lasting (up to 10 months) immunological onslaught, why Long Covid patients all experience lasting and varied symptoms (many have high levels of HMGB1 for months).

It took a few years for a research group to provide evidence that SARS-CoV-2 Spike Protein directly interacts with HMGB1, and thus, I suspect that for some of my recent hypotheses, it shall be the case. I believe this Spike Protein-HMGB1 complex is what enables Spike Proteins to get inside cells inadvertently and especially when interacting with other proteins, lipids, and sugar in the body, it can induce enough changes to shield the fragments from proper processing, thus Spike Protein can last abnormally longer.
Some authors even argued that HMGB1 can directly or indirectly modulate the levels of Bifidobacteria. I recently wrote about the effect of SARS-CoV-2 on Red Blood Cells, especially how it contributes to the formation of microclots and how it cascades from there, and secondarily, the effects of the infection on the long-term formation of RBCs. HMGB1 mediates such alteration. HMGB1 can mediate the formation of microparticles in the blood, altering blood coagulation, and induce the formation of both micro and normal clots.
There are other unconventional (non-canonical) effects that HMGB1 enables. It can interact with G-Quadruplex, altering how proteins are produced, and it can bind to DNA. Hypothetically, HMGB1 can act as a stealth gene delivery system, altering gene expression by epigenetic change, causing lasting, but easy to miss, changes in the body.
It is also one of the main players in aging.
• Senescence onset is accompanied by elevated extracellular HMGB1, including the reduced isoform (ReHMGB1), at cellular and organismal levels
• ReHMGB1 induces paracrine senescence in fibroblasts and propagates systemic senescence in vivo.
• ReHMGB1 enhances senescence phenotypes via activation of RAGE-mediated NF-κB and JAK/STAT signaling pathways.
• Reducing circulating HMGB1 levels alleviates systemic senescence and inflammation, and enhances muscle regeneration in mice.
• ReHMGB1 may serve as a systemic SASP factor and a potential target for age-related pathologies.
Extracellular ReHMGB1, but not its oxidized form, robustly induced senescence-like phenotypes across multiple cell types and tissues. Transcriptomic analysis revealed activation of RAGE-mediated JAK/STAT and NF-κB pathways, driving SASP expression and cell cycle arrest. Cytokine profiling confirmed paracrine senescence features induced by ReHMGB1. ReHMGB1 administration elevated senescence markers in vivo, while HMGB1 inhibition reduced senescence, attenuated systemic inflammation, and enhanced muscle regeneration.
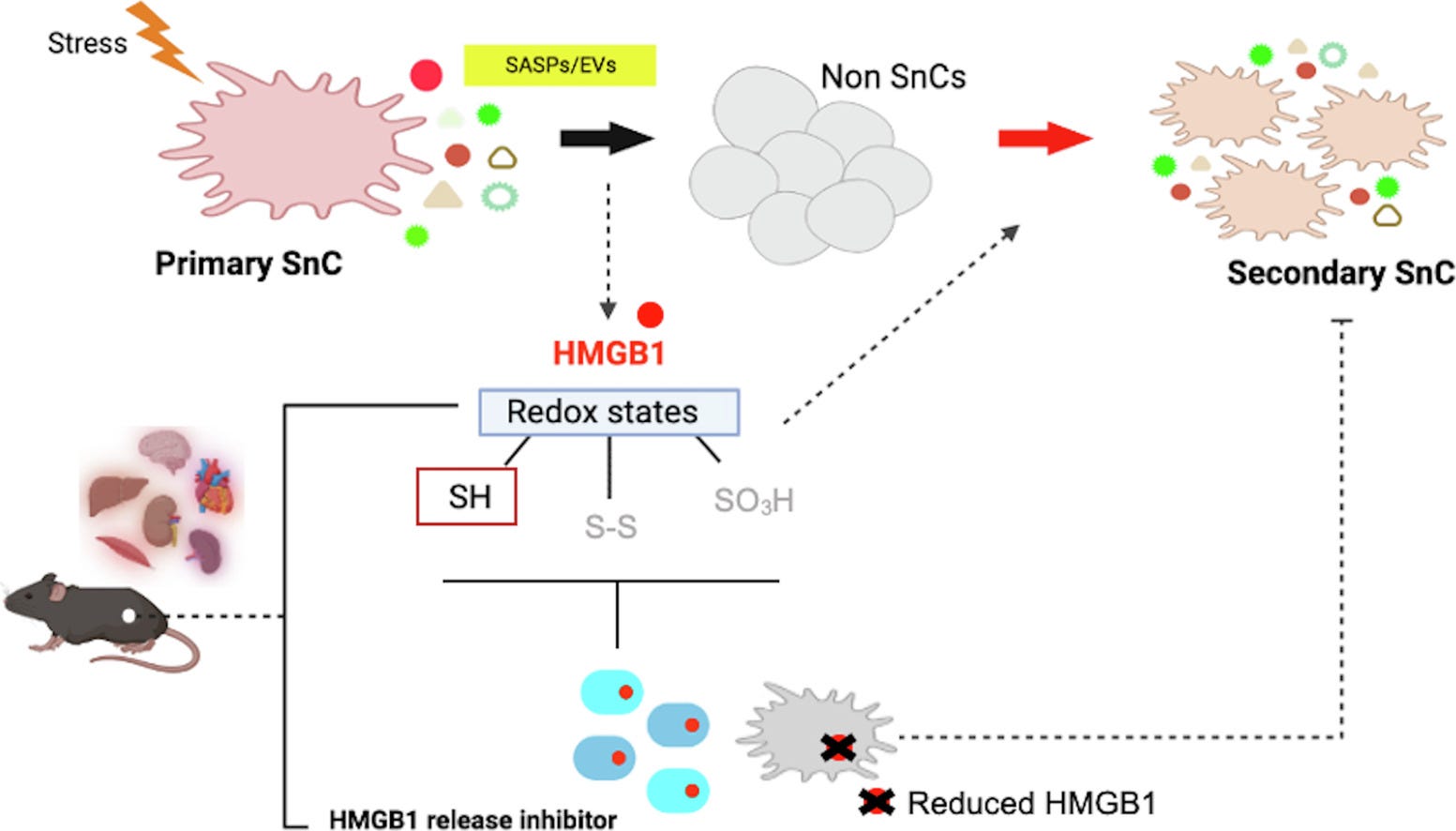
A hallmark of aging is the systemic decay of the immune system, fueled by chronic, low-grade inflammation, a state often called "inflammaging". As we age, our cells become senescent, commonly referred to as zombie cells, they stop dividing but refuse to die, instead pumping out inflammatory signals known as the Senescence-Associated Secretory Phenotype (SASP), and one of the most well-studied SASPs is HMGB1.
The researchers found that the specific, reduced form of HMGB1 acts as a "systemic SASP factor." When they administered it to young mice, it propagated senescence throughout their bodies, as if aging were a contagious disease. It triggered the same RAGE-mediated pathways we just discussed, leading to a cascade of inflammation and cell cycle arrest. Conversely, when they inhibited HMGB1 in old mice, they saw a reduction in systemic senescence, less inflammation, and even enhanced muscle regeneration. HMGB1, released by senescent cells, doesn't just signal aging; it actively drives it, turning a local problem into a body-wide crisis.
There is a remarkable similarity and shared pathways in this paper that demonstrate HMGB1 being a central player in senescence and aging, the deceleration of healing, and SARS-CoV-2, which I named The Great Accelerator years ago.
Consider becoming a paid subscriber, and if you are one, thank you !

Typical of me to miss fixing the title after days of research and hours of writing.
It's all so tiresome 😆
Professor - Another capolavoro.
"The goal of the researchers from the start here was to determine if HMGB1 could bind with HMGB1."
It was late and you were tired.
"It is also one of the main players in aging."
Indeed, in spades and how might we accelerate that process for billions of recipients?
It was known LONG AGO that:
1. N-linked glycosylation plays a crucial role in the secretion of HMGB1 AND in the structure and function of viral proteins, particularly the HCoV spike (S) glycoprotein.
2. The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming.
3. HCoV spikes create a redox shift phenom and increase HMGB1 which involves autophagy machinery and vesicular trafficking. Yet, as opposed to mere spike presence, the infection can cause a REDUCTION in autophagy. Oh the irony of it all?
A combination of the above can cause amongst other things - cellular senescence on a grand scale. Not to mention HCov's historically nasty habit of CNS invasiveness.
You continue to fill in many of the blanks admirably, keep plugging Mr. Mulder.