

Two caveats before delving into the article. First, cell lines lack an immune system, experiments with them are crucial, but shouldn’t be looked at from a cataclysmic perspective.
The second caveat is that most of the current and latest evidence points towards the failure of “misfolded protein” clearance being the problem, not merely the build-up. And these proteins serve more than one purpose.
Summary of both articles discussed here.
The protein that creates the infamous Amyloid Beta helps SARS-CoV-2 enter cells
The viral infection for long periods increases the levels of bad amyloid in the brain
The Spike Protein is most likely the switch that enables the inflammatory immune response responsible for this, it also directly interacts with the precursor
Some Long Covid patients have distinct muscle types different from “healthy” people
They have poorer oxygen transport, poorer mitochondrial health, more oxidative stress
They also have higher amyloid deposits and more immune cells infiltrating the muscle causing more damage
As I wrote the last time, fate or the universe aligned, and multiple papers were published recently that are significant. I didn’t plan to write this article so soon, but a recently published paper that fits the theme was published on January 4, so here we are. First things first.
First a little explanation on the (now) infamous protein we will be talking about. Amyloid Precursor Protein (APP), as the name implies, it is the precursor form of the infamous, “neurotoxic” protein Amyloid Beta. If you recall, Amyloid proteins play a role in the brain, they are functional and sometimes protective roles, and so does its precursor protein, playing a similar part.
For simplicity, APP is a big protein, and proteases (enzymes that cut bigger proteins into smaller blocks) will cut the APP into its smaller forms, one of them being the infamous Amyloid Beta. The authors raise good points in their introduction section.
Amyloid precursor protein facilitates SARS-CoV-2 virus entry into cells and enhances amyloid-β-associated pathology in APP/PS1 mouse model of Alzheimer’s disease
is a single-pass transmembrane protein abundantly expressed in the brain and metabolized in a rapid and highly complex fashion by a series of sequential proteases, including the ADAM10, ADAM17, β-site APP cleaving enzyme-1, and γ-secretase complex, which also process other key regulatory molecules
Moreover, the large ectodomain of APP, which has recently been revealed to be multifunctional, plays receptor-like roles.
A single-pass transmembrane protein is a protein that passes (duh) only one time throughout the cell membrane, contrary to other proteins doing it multiple times. This effectively affects how they behave, what they can do, and how they interact with other proteins. Among all the proteases cited here, the most significant is Gamma (γ, the y-looking thing) secretase, which is a group of, and not just one “thing”, given that Gamma is one of the main contributors to the “bad” amyloid.
Ectodomain is fancy scientific jargon for something that is found outside the cell, it is what interacts with other proteins and starts many functions and some cellular cascades. Also, I was somewhat happy to read the following from the authors themselves, as I wrote multiple times in the last 2 and a half years. The receptor used decides the fate of the infected cell.
All these findings suggest that ACE2 is not working solely during SARS-CoV-2 infection and the host cells entry of SARS-CoV-2 is a multi-receptor process.

SARS-CoV-2 S protein highly binds to N-terminal APP
To see if there is an interaction between the Spike Protein and the N-terminal of APP (the N-terminal of a protein meaning the first part of the protein) using a test called Localized Surface Plasmon Resonance. The test consists of using nanoparticles to see how light changes its behavior when two proteins you are studying interact with each other (this is an extremely simplified version of the test).
They decided to also test the binding strength of ACE2 as a reference point, and as one would expect, the Spike Protein bindings quite strongly to ACE2, but unexpectedly the Spike binds to APP too, for reference the ACE2-Spike KD (binding affinity) was 34.9 and APP 46. The lower the KD, the higher the affinity, the binding between Spike Protein and APP tended to become less stable and the proteins started to separate after a short while. The interaction was further confirmed with two other distinct tests.

The next step was testing if APP enhances SARS-CoV-2 pseudovirus (a form of non-infectious virus so you can run experiments outside BSL3 and 4 labs in a safe manner, we wouldn’t want highly contagious variants escaping from a lab, am I right ? *pure sarcasm) in two cell lines that had overexpression, meaning more than the normal, levels of APP. Cell lines overexpressing ACE2 were infected by the pseudovirus, but for cells overexpressing APP the infectibility was strengthened.
Their next step was testing in mice, if there was involvement of APP in brain infection, and once again, they overly expressed it (overexpression, meaning making a living organism produce more of something is very useful to study the effects of said something, but does not entirely equate into our bodies). Similar to the cell line tests, they found brain infection aided by APP. By using a APP N-terminal blocking peptide called AY51 they found the brain infection was significantly reduced.
The author’s next step was to use human brain organoids (mini-brains created from stem cells) and by using a method to remove APP expression in the “mini-brain”, they found viral load with low APP expression was significantly reduced. And for last, they tested different models of “chronic infection” to measure if it enhanced Amyloid Beta levels.
They first measured in vitro in a cell line, the data suggest a remarkable increase in both forms of amyloid beta, Aβ1-40 (amyloid fibrils), and Aβ1-42 (amyloid plaques that are more pathogenic and neurotoxic). Infecting 2-month old mice used in Alzheimer’s research and observation was done at 4 and 11 months of infection.
Compared to the control group, as one would expect, the number of plaques was significantly increased, and at 11 months of infection, there was activation of astrocytes and microglia, denoting that long-term infection in the brain will induce inflammation, and as we would expect, accumulation of the infamous Amyloids.
The author’s conclusion speaks better than I could.
In conclusion, we have identified APP as a receptor protein for nerve cells and revealed a previously unknown mechanistic insight into COVID-19-related neuropathological sequelae. A systematical examination of multiple Omicron sub-variants on potential brain dysfunction would be warranted in future studies. The Spike protein could function as an immune switch to Aβ production and contribute to neurological changes in COVID-19 patients.
Protein misfolding and amyloids have been a massive personal interest of mine for the longest time, especially in regards to SARS-CoV-2 infection and that closing remark in bold letters instantly reminded me of another paper (so much that I checked if these were the same authors). The following is a complementary article to this one, and both fit extremely well together.
Spike protein S2 enhances amyloid-β production

A paper I didn’t think I would ever write about was published today, but before I went to write about it, I wanted the “receipts”. This is as far as I found it, my private (encrypted) chats with the person I collaborated with in the PAID hypothesis, therefore basically the
Before delving into the second, and very significant paper, I present you these and a old, perhaps helpful article.

Creatine supplementation improving Long Covid symptoms

I have been waiting for a while to write a bigger article on this subject, I wasn’t exactly holding back, but waiting for some level of good evidence, and this also adds to one observation I mentioned multiple times, to multiple people. Sometimes supplements take time to “
Muscle abnormalities worsen after post-exertional malaise in long COVID
Long Covid is a highly complex condition, and while for now we lack a formal classification of its many subtypes, it is clear there are distinct types of “long covid”. Here the authors used 25 well-definied Long Covid patients and controls and induced post-exertional malaise (PEM), commonly referred to as “covid crash”, they used both blood and skeletal muscle biopsies before and after the PEM (as someone who is a hyper metabolizer of any anesthetics except fentanyl, I felt the pain each biopsy I did…bruh moment here for you). None of the participants was hospitalized at any point due to SARS-CoV-2 infection, which demonstrates they did not develop a severe infection.
Limited exercise capacity in long COVID
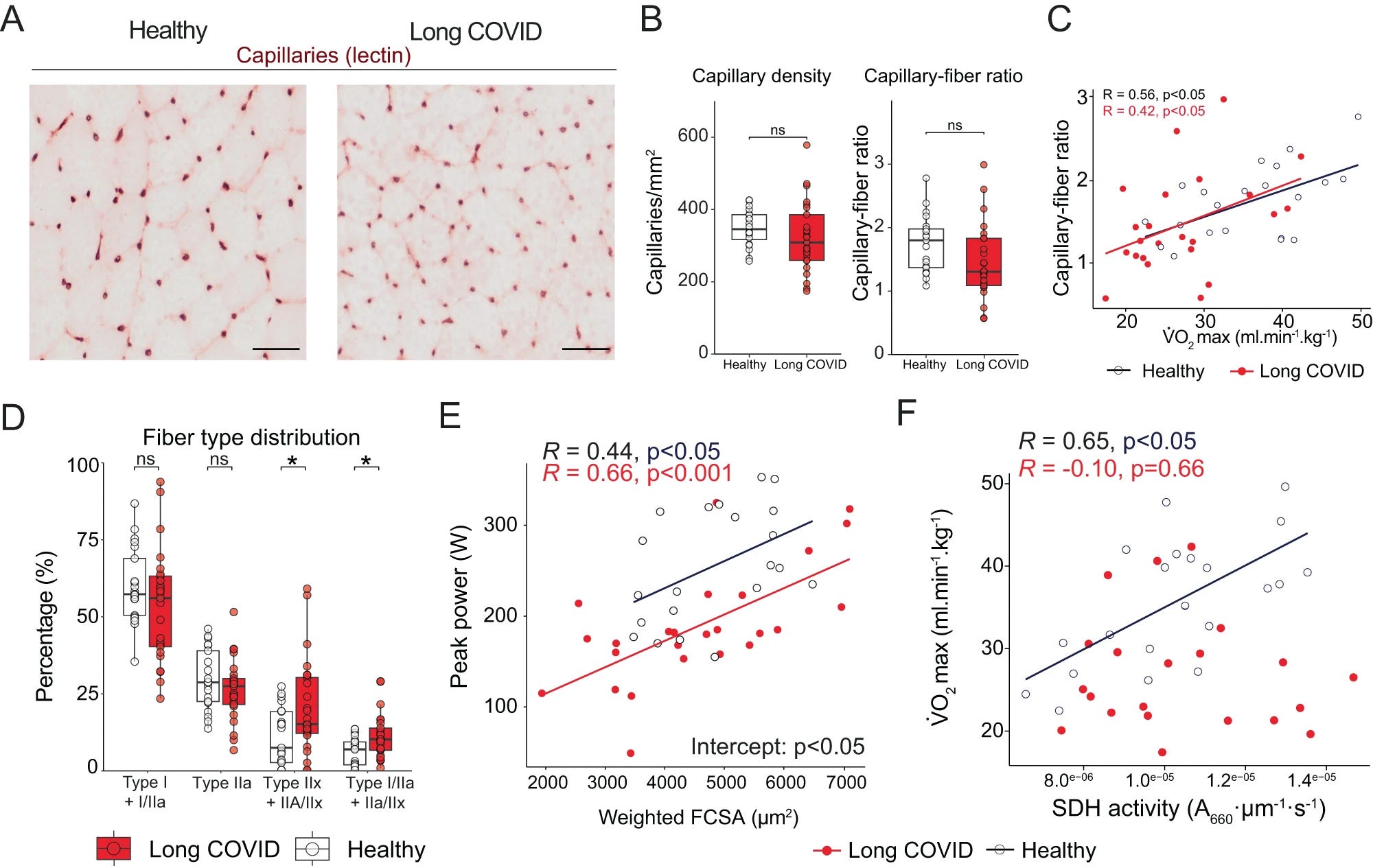
“All participants performed a cardiopulmonary exercise test on a cycle ergometer.” The tests found Long Covid patients had lower VO2max (a marker for cardiorespiratory fitness during exercise, measures the maximum amount of oxygen the body can use, one of the favorites of many health and gym influencers for longevity), had lower exercise intensity capacity, lower ventilatory function during exercise, they had lower gas exchange during exercise, lower oxygen transport, and utilization, lower peripheral oxygen extraction (muscles have a problem using oxygen), this indicates there is a level of impairment on lung ability to move air in and out of the blood. Lactate, a byproduct of cell metabolism, produced in abundance during exercises, did not differ between Long Covid and the control group.
The capillary density and the capillary-to-(muscle)fiber ratio were not significantly different between the two groups, but there was an observable trend towards lower capillary-to-fiber ratio in Long Covid. The LC patients had a distinct muscle difference, with more highly fatigable glycolytic fibers than controls, and female patients had less fatigue-resistant Type I fibers. LC couldn’t achieve the same “peak power” as controls because they had different muscle fibers at some level.
Lastly in this section, and most importantly, LC had lower oxidative phosphorylation capacity, suggesting lower mitochondrial function.
Metabolic dysfunction and post-exertional malaise
To understand the aspects contributing to the “crash” the researchers got a biopsy of the vastus lateralis (the big thigh muscle) before and one day after inducing PEM. All LC patients had symptoms following the exercise, muscle pain, greater severity of fatigue, and cognitive symptoms up to 7 days.
Oxidative phosphorylation (cells use enzymes to create a reaction and release energy from nutrients) was decreased after the exercise in both controls and patients. SDH (Succinate Dehydrogenase, an enzyme complex in mitochondria that helps create energy) wasn’t reduced in the control group one day after the exercise, but it was in LC.
Effectively means the mitochondria in the muscle cells of LC patients don’t seem to function as efficiently as they should, and there aren’t as many of them as expected.
To better understand the metabolic response behind PEM, they measure over 100 metabolites from muscle cells and over 80 from blood. Many of the most important metabolites in the TCA cycle (chemical reactions to generate energy inside your cells) were lower in LC patients.
Skeletal muscle creatine concentrations were lower
S-adenosylmethionine (SAM) was lower, indicating reduced methylation and SAM cycle
Dihydroxyacetone phosphate, important for lipid biosynthesis and glycolysis was reduced post-PEM
Hydroxyphenyl acetic acid was lower, showing increased production of ROS in the mitochondria
“Many amino acids were not different between groups at rest, but tended to be lower in patients upon the induction of post-exertional malaise”
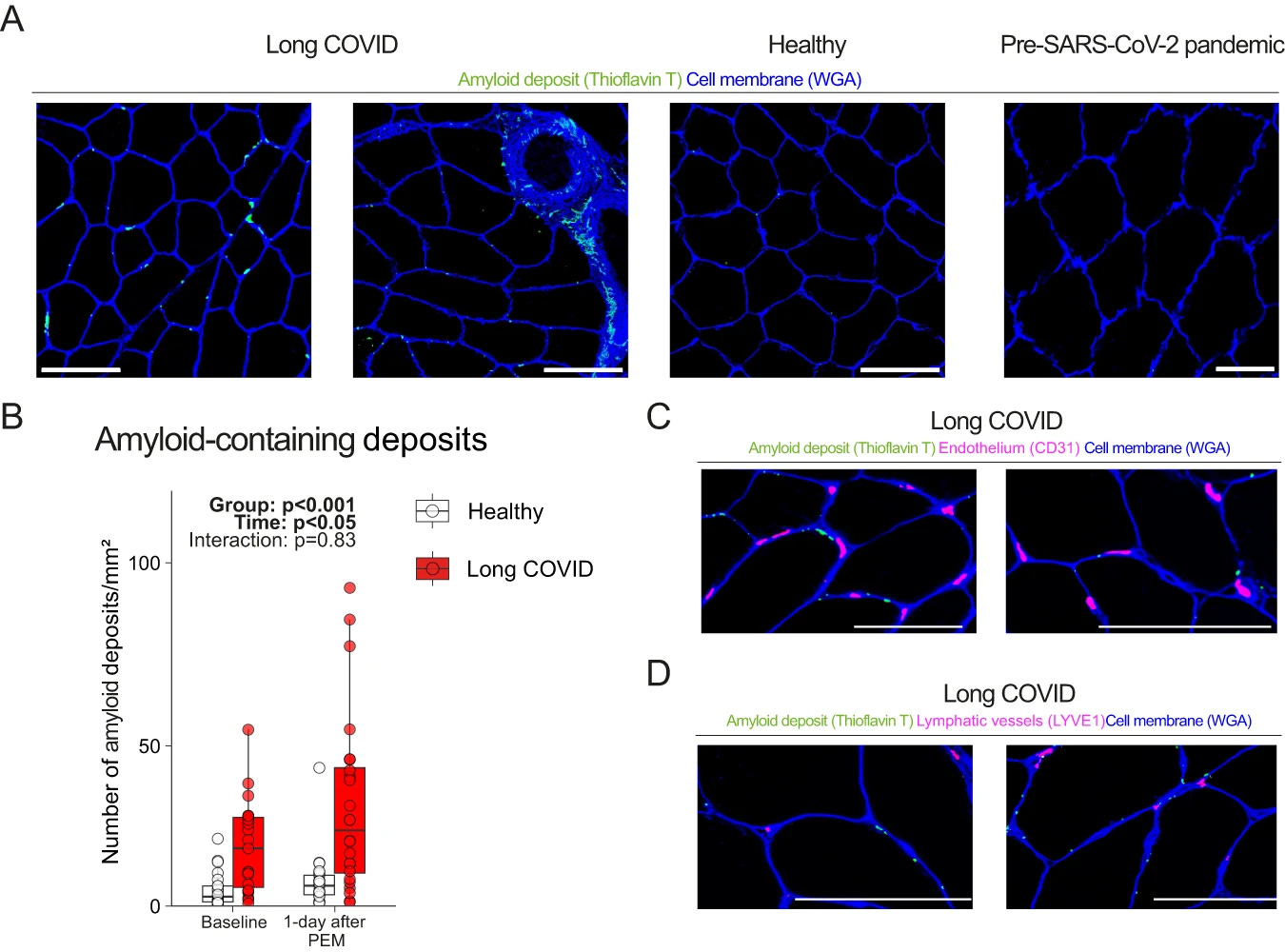
Exercise-induced amyloid-containing deposit accumulation in skeletal muscle
This group of researchers started this research 2 years ago, but bureaucracy takes a long-time, 2 years ago, amyloid-related mechanisms were quite in vogue, especially after amyloid fibrils-based microclots were found in some LC patients. So here they wanted to study if amyloid-containing deposits were present in the muscles of LC patients and if PEM changed the concentration.
They had biopsies obtained before the pandemic, and they used this as a double “control” group, comparing the levels pre-pandemic, with the levels of the control group (infected with SARS-CoV-2 but no LC), and pre and post had similar levels, but LC had a higher concentration of these amyloid patients at baseline, and increased equally on both groups.
Since the research was initiated a couple of years back, the authors decided to visualize the amyloid deposits with keen interest in the capillaries or lymph vessels and found none. They found the deposits next to the capillaries, and in between the muscle fibers, and this didn’t equate to localized hypoxia (one of the proposed mechanisms for the LC crash, micro clots in the capillaries leading to low oxygen in specific regions of the body).
I will get back to the following quote shortly.
“The underlying reason for the increased intramuscular accumulation of amyloid-containing deposits during post-exertional malaise remains elusive.”
Exercise-induced myopathy in long COVID
To understand the causes of increased muscle weakness, fatigue, and pain after exercise in LC patients they look into the biopsies. A larger percentage of LC displayed small atrophic fibers and focal necrosis, which increased significantly after exercise. Exercise in any person induces an inflammatory response and localized reaction, but in the case of LC they had exacerbated tissue damage.
Since muscle is a very “regenerative” tissue (plastic/plasticity is the correct term) the authors also looked into regenerative evidence, and regenerating fibers were seen in both groups, also at similar levels post-exercise, but the baseline in LC is higher
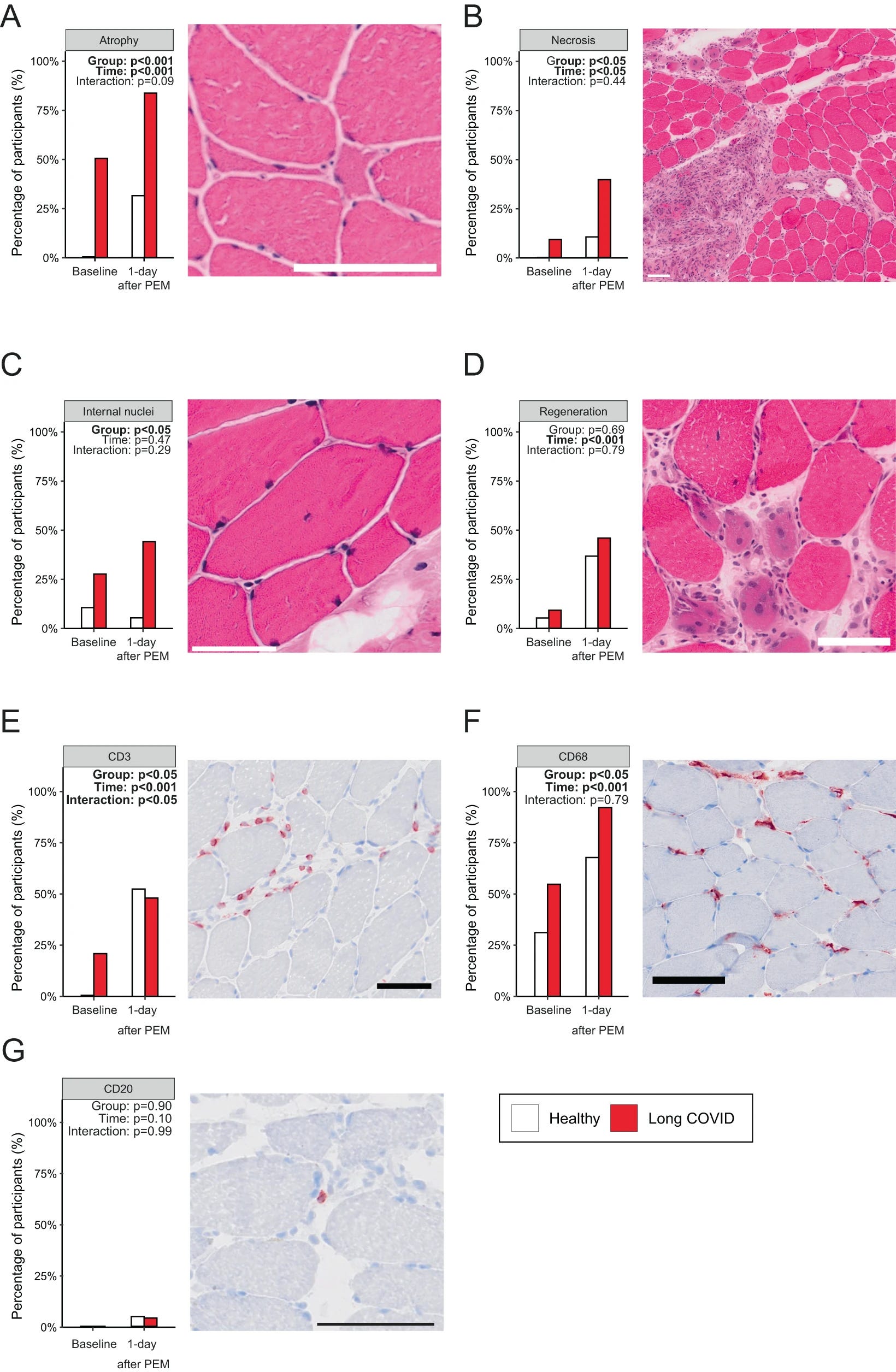
Since immune responses are part of exercise, especially exhaustive exercise, they measured immune cell infiltration (usually meaning immune cells moving from blood into somewhere else, therefore “infiltrating”), they observed that more LC patients had CD68+ macrophage infiltration in skeletal muscle compared to the control. Exercise caused a similar increase in infiltrating macrophages in both groups.
T-cells (CD3+) were absent in healthy controls, but present in LC before exercise. The exercise-induced accumulation of these T-Cells, but this response was less pronounced in LC patients. B cells were not different between both groups before or after exercise. Lastly, given the evidence they reasoned the infiltration of immune cells could be caused by mitochondrial DNA, which is highly inflammatory and can act as a signaling molecule to trigger systemic inflammation, but they found none.
They also found no increase in muscle breakdown produces, such as creatinine or creatine kinase in both groups. Creatine kinase especially is one of the “easiest” tells for Amyloid Myopathy, a rarer form of localized Amyloidosis (which is systemic). This means this is not (and never was) amyloidosis, nor it ever be except for a handful of outliers.
Cortisol was also the same between groups. One of the proposed causes for many of the dysfunctions presented in Long Covid and much of the damage post-severe disease is the viral fragment persistence (one that I proposed too). In the author’s own closing remarks
Further studies into the composition of these amyloid-containing deposits and the possible role of entrapped (auto-)antibodies43 in relation to post-exertional malaise and long COVID are warranted.
The mere presence of SARS-CoV-2 nucleocapsid protein in skeletal muscle is unsurprising, as nucleocapsid protein can be present up to a year after infection in blood49,50. It is however unknown if the full virus is present, or only protein remnants. Because the spike protein is located on the exterior of the virus, this protein may have different proinflammatory/coagulatory effects compared with the nucleocapsid protein. The absence of clear distinctions in the quantity of nucleocapsid protein and the equal presence of B- and T-cells following exercise suggests that factors other than viral persistence are associated with the pathophysiology of post-exertional malaise in patients with long COVID.
This is a remarkable finding because it has been proposed for a good couple years now that the Nucleocapsid protein has amyloidogenic properties (meaning it can form the infamous amyloid). Here the Nucleocapsid protein presence didn’t equate to the deposition of amyloids in the muscle.
As I have said repeatedly, it is very easy for me to point X, Y, or Z and ask researchers to do 60 different lines of research, because I am sitting on my butt, and I am not running the experiments, which are very time-consuming (especially if there are patients involved).
However, I would love for some group to analyze or sequence the contents of the amyloid deposits, especially for Human Endogenous Retroviruses. I also suspect at least in some, that amyloid deposition is a defense mechanism to stop tissue inflammation and degradation, a stop-gap as the body attempts to lower the inflammation and achieve the recuperating threshold necessary for healing.
I planned to write other articles, but the next one will be a follow-up on this one since it fits both papers discussed here and links other aspects of all of this. May take a few days.
If you chose to support my work, thank you very much !

i stand and applaud from the aisles, appreciate the graphics for their colorful beauty alone, as I cannot quite follow all the scientific meanings and implications. Real logic rings true. And the blue and black images look like snakeskin tattoes, just sayin. Also, like bee hives...I am made of both, apparently...
Hey it’s been a while. I think covid is synergising with HPV and perhaps was designed to tag team with many peristaltic viruses from the start, on its own, bad but perhaps not obviously oncogenic but when paired up with other common viruses. BOOM. It may even turn low risk HPV subtypes to high risk.