


SARS-CoV-2, biofilms and co-evolution
"Accelerated" evolution dare I say

Life (or fate) has interesting twists and turns, and lately, I have been glad that sometimes I am forced to postpone a Substack I planned to publish. First, I will write a short simplified summary, but for a better and more profound overview of everything we are about to go through, these Subsatcks are a “must read”.



The recent substack about SARS-CoV-2 Nucleocapsid Protein and protein misfolding would be a good read too.
Simplified Summary
SARS-CoV-2 molecular structure and its composition (the amino acid sequence) has many unusual, “novel” features, the Furin Cleavage Site being the one most commented on in the last 3 years. Among its many features, a few bear more weight not only by how unique they are, but what they do.
SARS-CoV-2 Spike Protein shape and amino-acid sequence create a peculiar unique shape that can both pierce cellular membranes, meaning the Spike can bypass how cell often function by using receptors, and just enter a cell, often leading to that cell’s death
The same effect can also “skin” bacteria and use that “skin” to become an inflammatory bomb (LPS, Endotoxin)
The same structure and effect can also affect biofilms, deconstructing their structure (bursting) and releasing their contents
The virus does this, and the Spike Protein by itself does this
Among dozens of the Substack, I have written on SARS-CoV-2, the ones referred to here bear the brunt of the weight into significance. Finally, a group of researchers wrote a proper hypothesis on SARS-CoV-2 and biofilms.
Biofilms possibly harbor occult SARS-CoV-2 may explain lung cavity, re-positive and long-term positive results
Multispecies biofilms exist both externally and within the host in nature (Von Borowski and Trentin, 2021). Fahrenfeld et al. detected the N1 and N2 genes of SARS-CoV-2 in sewage biofilms, which can be considered as a vitro model (Fahrenfeld et al., 2022). Actually, biofilms with viruses have been reported to exist in multiple microbiotas. Multispecies biofilms which encompass Gram-negative bacteria and filamentous fungi and enteric viruses have been spotted, as well as Candida albicans biofilms comprised with the coxsackievirus type B5 (CVB5) or herpes simplex virus 1 (HSV-1). Gomes’s team confirmed the presence of SARS-CoV-2 in oral biofilm samples (Gomes et al., 2021; Von Borowski and Trentin, 2021). In this paper, we hypothesize the possible mechanism of the long-term retention of SARS-CoV-2 in the form of biofilm which leads to lung cavities, re-positive and long-term positive nucleic acid tests
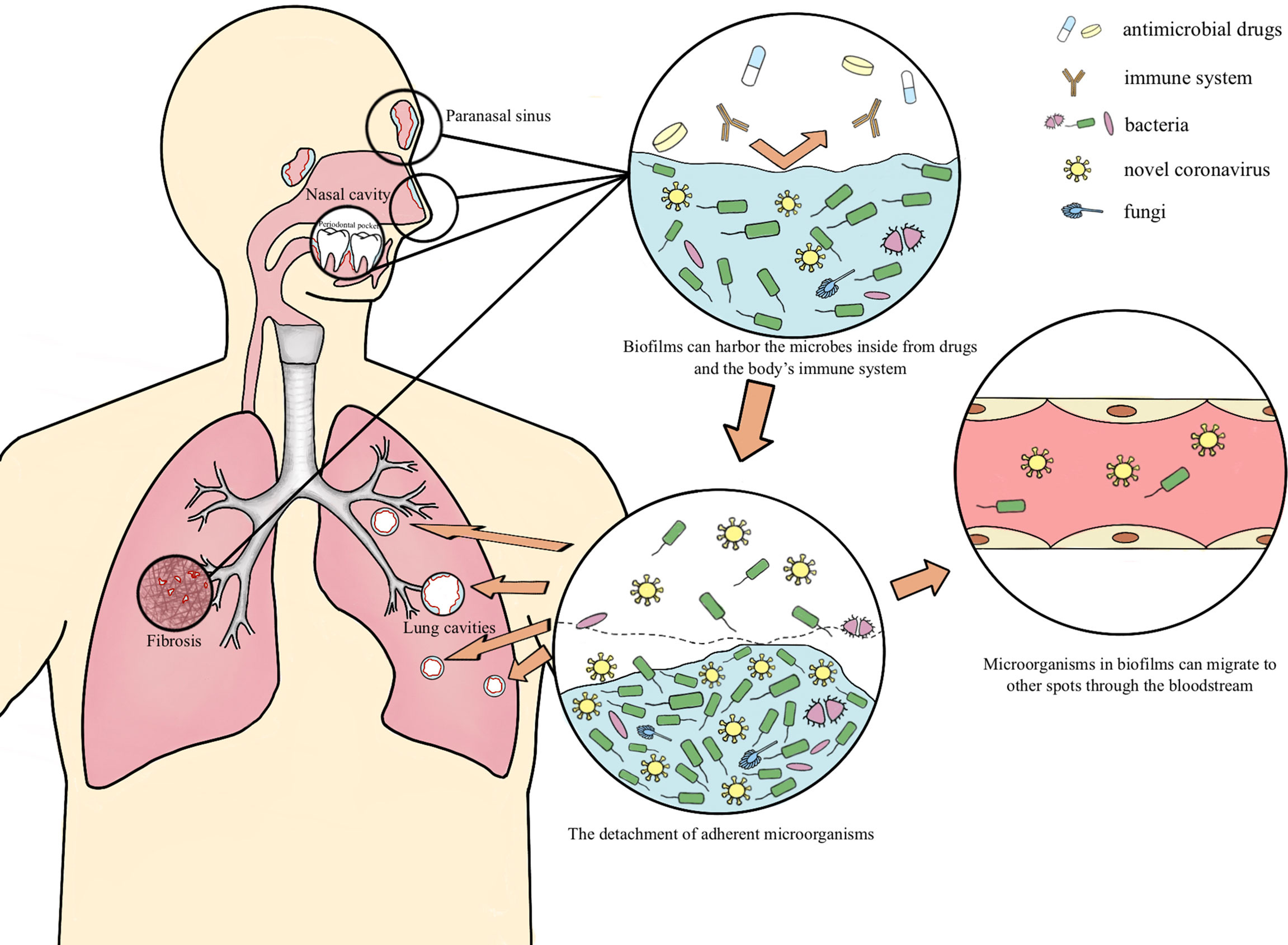
This entire paper is full of pertinent references to observations by other researchers and clinicians, and worth checking out for a (almost endless) rabbit role of connections. Co-infections were, are, and will be for the most part the causative “agents” of damage, morbidity, and long-term damage, both acutely and immune-mediated at low-grade inflammatory levels. Many of the “treatments” and comorbidities present in different SARS-CoV-2 infections are either byproducts of or aid in the infection of other pathogens, such as immunosuppression, cardiovascular disease, diabetes, mechanical ventilation treatment, long-term antibiotic use, glucocorticoid therapy all influencing co-infection outcomes.
Many of the pathogens present in co-infections such as M. pneumoniae, P. aeruginosa, H. influenzae, and K. pneumoniae, Streptococcus, Klebsiella, and Staphylococcus can create and live inside biofilms. The author goes to proposed that the lung damage caused by SARS-CoV-2 and co-infection with other pathogens is able to create the right environment for the appearance of biofilms and that biofilms are also present in the nasal mucosa of certain individuals (sinusitis) and the fact that dental biofilms harboring SARS-CoV-2 exist. To this last one, I actually know half a dozen people who got “re-infected” because the dentist unawarely bursted biofilms harboring the virus. More significant than biofilms in these regions, are the other regions biofilms form, rarely in the circulatory system, but the most common and significant is the gut.
Currently, there has been quite a large debate among researchers on why/how certain patients keep testing positive (referred to in the paper as re-positive) for months to no end, with many potential explanations being presented, from failure to clear the virus (viral persistence), failure to clear virus parts (viral fragments) and now biofilms harboring latent virus and I would personally at viral fragments too.
SARS-CoV-2 likely interaction and stealth ability (hiding inside biofilms) would go a long way to explain many of the paradoxical dynamics and molecular pathways present in Long Covid and much of the long-term sequelae from the infection. Biofilms are not inert “houses of bacteria and fungi” but active biological components, that elicit immunological and inflammatory responses, and they even have residual endotoxins in their structure. These mechanisms could also explain its bacteriophage behavior.
These interactions, and manipulation of biofilms can also explain a few mechanisms in regards to Long Covid that I still need to properly write about. Candida, one of SARS-CoV-2 best friends can “entrap” Herpes virus on its biofilms and consequentially the virus will avoid the immune system and drugs, herpesviruses such as EBV and CMV both now known to play a significant role in certain Long Covid groups, can also influence periodontitis (dental biofilms, remember). A multi-prong attack, it is not merely SARS-CoV-2 causing momentarily immune suppression, but actively releasing pathogens from biofilms.
I will attempt to summarize the following two papers for brevity and simplicity's sake, but I do recommend anyone to read both, especially the second. More than once.
The Staphylococcus aureus protein IsdA increases SARS CoV-2 replication by modulating JAK-STAT signaling
Established an in vitro SARS CoV-2-Staphylococcus aureus co-infection model
The S. aureus protein IsdA increases SARS CoV-2 replication
IsdA enhances the production of SARS-CoV-2 infectious virus particles
IsdA manipulates host JAK-STAT signaling to the benefit of SARS CoV-2
One significant complication of respiratory viral infections, including CoV-2, is increased susceptibility to secondary bacterial infections. Although frequency of co-infection varies between reports and locations, the overall rate in the general population is roughly 5%.9 However, incidence jumps to 25–30% of hospitalized patients, and is as high as 40% in intensive care unit (ICU) patients. Importantly, the mortality of CoV-2 and bacteria co-infected patients can be as high as 35%, despite the almost universal administration of antibiotics.
Data from patients infected with CoV-2, and with laboratory confirmed bacterial co-infection, show the most commonly isolated species to date has been the Gram-positive opportunistic pathogen S. aureus.
Right now most of the world is fully aware that co-infections were the actual “killers” and slowly we are finding evidence that co-infections also drive parts of the whole dysfunction, but this was a first. One of the most common co-infections with SARS-CoV-2, also a common infection post-vaccination and one of the most studied biofilm organisms in recent scientific memory is found here to affect viral replication.
By meticulously testing different steps of co-infection, such as if Staph. A. could infect cells as easily with or without prior SARS-CoV-2 infection, and bacterial invasion, they first found that the presence of SARS-CoV-2 didn’t affect bacterial replication, but Staph presence increased the number of infectious SARS-CoV-2 particles in the cells. Followed by testing if the mere presence of Staph cells alone dead or alive could impact the growth of SARS 2, finding that online the live bacteria can induce such growth, testing other strains, so this isn’t strain specific such as many other particular cellular effects of many pathogens, SARS-CoV-2 being a perfect example of this effect.
Based on the observation of using different strains of Staph A. and said strains regardless of virulence status demonstrating pro-viral effects, the researchers hypothesized that perhaps one or more proteins released by Staph. are the effectors of this atypical function. By using bacterial mutants lacking specific proteins they found that all of them, except the mutant lacking Sortase A (srtA), kept their pro-viral effect. Sortase A is an enzyme responsible for anchoring a group of proteins known as “cell-wall anchored” (CWA) proteins to the bacterial cell surface.
Without Sortase A, CWA proteins were still produced but directly secreted into their testing method, eliminating Sortase A partially eliminated the pro-viral effect, meaning proteins that are anchored by Sortase A rather than the protein itself enhanced viral replication, with finally founding that IsdA was the one responsible for this effect. IsdA alone didn’t have such a pronounced effect so its pro-viral activity is more significant when the protein is shed from the bacteria, with the authors arguing that it probably attaches itself to other peptidoglycans, sugar components of cell walls.
After RNA sequencing to understand how this occurs at a protein/molecular level, authors found that IsdA from Staph. A. modulates JAK2-STAT3 signaling to enhance SARS-CoV-2 replication. At higher levels IsdA itself affects STAT3 therefore Staph. A. targets host cells via IsdA and this pro-viral effect is an uninteded byproduct.
JAK-STAT is one of the most important pathways in our bodies, JAK received signals from outside the cell and deliver them to the STAT, when STAT proteins are active they turn on specific genes and proteins in the nucleus of cells.
These dynamics are important because as I wrote quite a few times, a lot of the damage observed from the initial viral infection be it short or long-term, and the displacement of other seasonal viruses and surge in different types of infections were initiated by a “first salvo”, a complex relationship between SARS-CoV-2 and many other common bacteria and viruses. The body enters a cascade (doom loop) attempting to reach homeostasis again, this specific mechanism is one of the cores of the failure and outstanding damage of the mRNA platform.
Before delving further, I highly advise you to read the next paper in its entirety. Whatever I will write doesn’t make justice to the paper. The additions might add some value, but the paper itself is remarkable.
Candida albicans-enteric viral interactions—The prostaglandin E2 connection and host immune responses
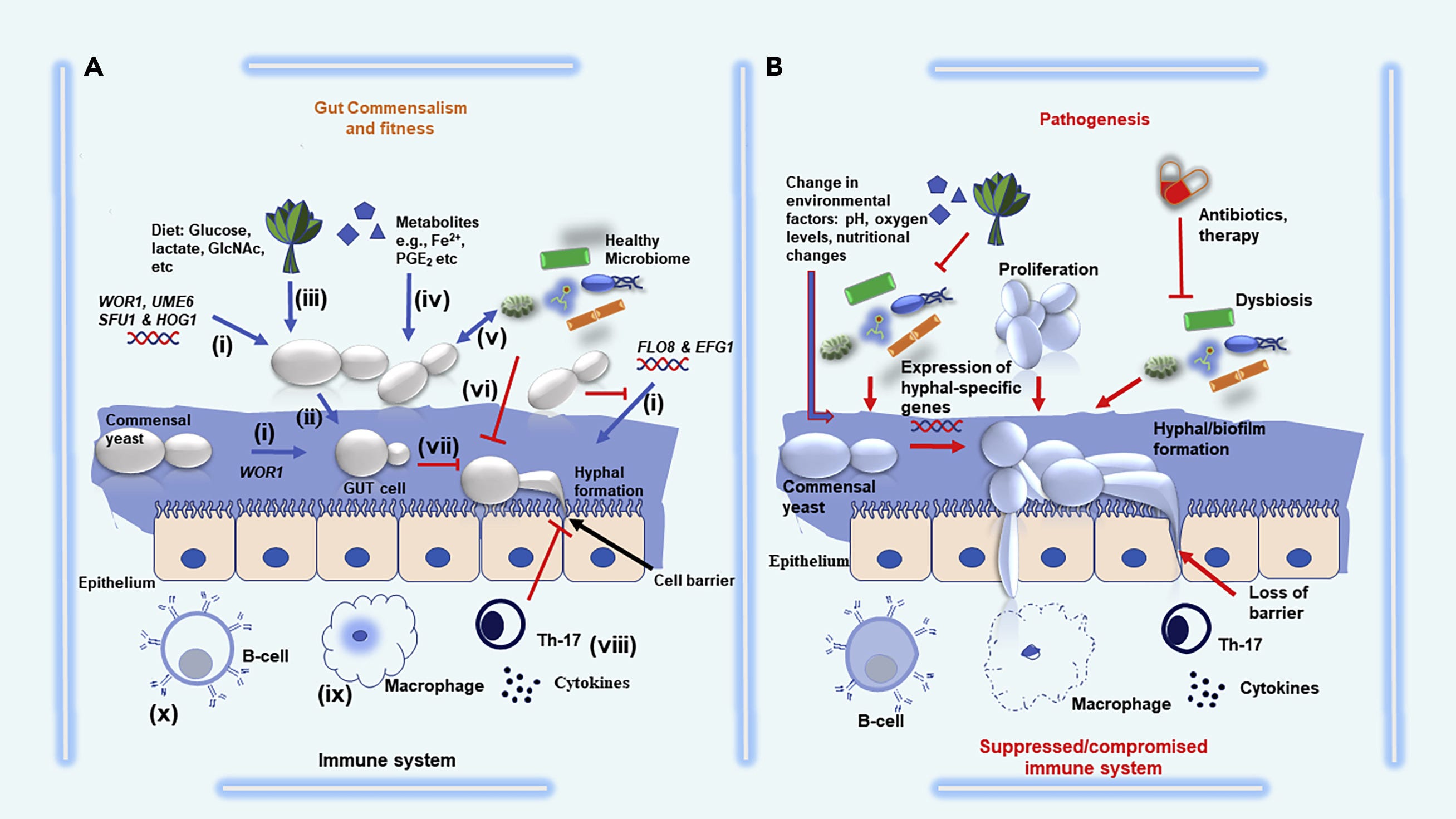
The ability of Candida albicans to adapt its metabolism is crucial for successfully colonizing the mammalian gut. In the gut, where the preferred carbon source, glucose, is limited, Candida albicans show flexibility by utilizing alternative carbon sources such as lactate, citrate, or glycerol. Interestingly, Candida albicans lacks certain sites that would typically inhibit the use of alternative carbon sources, allowing it to utilize these sources even when glucose is present. Mutant strains that cannot use alternative carbon sources have reduced competitiveness in colonizing the gut compared to normal strains.
The regulation and acquisition of iron by Candida albicans are important factors in its colonization and adaptation to the iron-rich gut environment. In different environments, Candida albicans adjusts its iron-uptake mechanisms based on iron availability to support its colonization and overall fitness in the gut.
It's worth noting that Candida albicans use arachidonic acid (AA), derived from the host, to produce prostaglandin E2 (PGE2), a lipid inflammatory mediator. Initially associated with virulence, PGE2 production contributes to Candida albicans' ability to colonize and compete in the gut.
Candida albicans forms biofilms on multiple surfaces such as catheters, medical implants, and host mucosal surfaces, contributing to high resistance to antifungal drugs (e.g., fluconazole and amphotericin B) and a high mortality rate.
Alterations in environmental factors such as a shift in pH, oxygen levels, diet or use of antibiotics, and immunosuppressive drugs can promote biofilm formation and over-proliferation of C. albicans
The interactions of C. albicans biofilms and viruses may also have a reciprocal effect on the growth and establishment of C. albicans biofilms.
For example, HSV-1 infected macrophages (derived from THP-1, human acute monocytic cell line) show enhanced phagocytosis of C. albicans but are impaired in intracellular deactivation of the ingested fungus and ultimate antifungal activity.
The study showed that HSV-1 downregulates toll-like receptors (TLRs), TLR-2 and TLR-4, which are essential in host recognition of C. albicans infections, suggesting that HSV-1 presence favors fungal survival and immune evasion and may contribute to disease progression.
In another study, HSV-1 and HSV-2 were shown to enhance adherence of C. albicans to HeLa cells in a virus dose-dependent manner, indicating a possible interaction of both pathogens even at infection sites.
Candida Albicans is known to colonize many surfaces and is responsible for a good portion of hospital-acquired infections, and while its biofilm interactions with viruses is a recent research avenue some evidence points towards the reciprocal effects Candida can have with certain viruses, such as HSV-1 downregulating TLRs, the receptors essential for recognizing different type of pathogens, also yes, this is the very same Herpes mentioned at the begging of this piece. In regards to Herpes-1 Candida has multiple quasi-symbiotic effects, and it would be “safe” to deduce based on interdisciplinary evidence it will do the same with other viruses.
Candida albicans also produces PGE2 from host-derived AA through the activation of phospholipase A2 which hydrolyzes glycerophospholipids at the sn-2 position.
PGE2 production by C. albicans is upregulated during biofilm formation and is essential for competitive fitness during gut colonization. In addition, PGE2 produced by C. albicans biofilms stimulates the growth and biofilm formation of pathogenic bacteria such as Staphylococcus aureus in dual-species biofilms.
It is also well established that PGE2 interaction with enteric viruses can enhance viral replication, and this activation may ultimately exacerbate the outcome of the disease.
This raises the question of how PGE2 would affect the outcome of the possible C. albicans-virus interactions during polymicrobial gut colonization, biofilm formation, or multi-species infections. Here we highlight some enteric viruses that utilize PGE2 or initiate expression of the COX/PGE2 pathway for viral replication, which may ultimately influence the unexplored C. albicans-viral interactions, especially during gut colonization or in polymicrobial gut infections.
By fooling the body to produce arachidonic acid (an omega-6 “fat”), Candida sequesters AA and produces its own PGE2, and PGE2 produced by Candida both stimulates biofilm formation, and growth of Staph. A. Yes, the same Staph mentioned above, creates a dual-species biofilm.
Now why am I bringing the Candida paper and its multi-faceted dynamics here ? Not merely because the Spike directly interacts with Candida biofilm formation, or Candida and Staph forming a symbiotic relationship, or all mentioned above, but because Candida has been observed as playing a significant role during and post-infection. This 2022 paper about mRNA-LNP found that the mRNA-LNP reprogrammed the immune system in a way that completely shifted the response to certain pathogens, with Candida Albicans responses decreasing, meaning people vaccinated with the platform are at a higher risk of Candida infections, and so does severe infection with SARS-CoV-2.
Candida colonization has profound long-term effects on populational health, such as affecting the incidence of Sjögren's Syndrome, may play a role in the development of Celiac disease, Candida and Staphylococcus Aureus both will induce a Th17 biased response that will exacerbate autoimmune diseases, such as kidney disease, there is more abundance of Candida in Multiple Sclerosis patients. The complexity of the microbiome, SARS-CoV-2, and bacterial and fungal pathogens can’t be understated, in the last few months, we now have a decent initial picture of how many pathways are crossing and creating symbiotic pathogenic cascades.
And just for fun, while biofilms are made of many components among the most common are carbohydrates.
Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses
Human T cell leukemia virus type 1 (HTLV-1) is a lymphotropic retrovirus whose cell-to-cell transmission requires cell contacts. HTLV-1–infected T lymphocytes form 'virological synapses', but the mechanism of HTLV-1 transmission remains poorly understood. We show here that HTLV-1–infected T lymphocytes transiently store viral particles as carbohydrate-rich extracellular assemblies that are held together and attached to the cell surface by virally-induced extracellular matrix components, including collagen and agrin, and cellular linker proteins, such as tetherin and galectin-3. Extracellular viral assemblies rapidly adhere to other cells upon cell contact, allowing virus spread and infection of target cells. Their removal strongly reduces the ability of HTLV-1–producing cells to infect target cells. Our findings unveil a novel virus transmission mechanism based on the generation of extracellular viral particle assemblies whose structure, composition and function resemble those of bacterial biofilms. HTLV-1 biofilm-like structures represent a major route for virus transmission from cell to cell.
In one of my favorite television series, one of the protagonists said “Time is a flat circle”. If time is a flat circle, SARS-CoV-2 is like time.


If you chose to support this substack or used Kofi as a one-time thing, or shared this Substack, thank you, appreciate the support.
Excellent John, an area little discussed, possibly as it crosses specialisms. As with cancer disease begets disease & coinfection in a downward spiral.
I’m only half way thru this and I almost jumped for joy!
This is the link I’ve been looking for!!!
Two of my children have been plagued by autoimmune issues ever since “recovering” from Covid. One developed alopecia and the other started having idiopathic anaphylaxis. We’ve been to immunologists and allergists to no avail...they’re all baffled as to why my healthy kids with no prior history of immune issues would be suffering spontaneously from major inflammatory responses.
All along I’ve suspected that Covid was indirectly responsible. It’s the only thing that’s “changed” in the last 2 years in our life.
I will read the rest of this and work of the courage to bring this all to the hematologist I’m set to take my 15 year old to in June. I’m sure the doctor will think I’m nuts. But our family nurse practitioner has said that she’s been giving referrals to hematology and allergy like crazy lately....so many mysterious ailments.