


Spike Protein Impairs Mitochondrial Function in Human Cardiomyocytes
Mitochondrial Dysfunction and the Kynurenine Pathway

It has been a few days since the last one, hasn’t it ? In case my readers are not aware, my aunt and cousin came to visit from overseas, and it has been quite a long time since the last visit. I don’t have a problem sharing parts of my life here, so the reason I spent a few days away is that I owe a lot to her, especially in my early life, when I was severely poor. As in no food poor, no paper to write poor.
Normal activities resume today. I do advise you to take days off the internet too, to clean your brain from the information deluge. This time “away” was also fortuitous, because quite important papers were published.
Interestingly, in line with the last published substack, we circle back to what I mentioned before, mitochondrial dysfunction is at the center of most problems in regards to the virus or the Spike. I have many pieces written about it, but the “most important” ones would be the following ones, with the second, named Part IV tieing a lot of other topics we covered so far together.

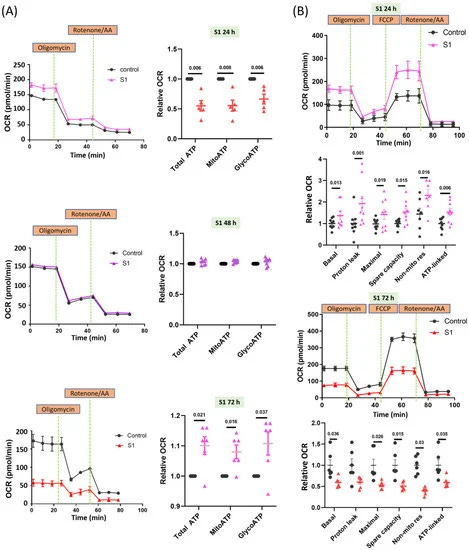
Spike Protein Impairs Mitochondrial Function in Human Cardiomyocytes: Mechanisms Underlying Cardiac Injury in COVID-19
COVID-19 has a major impact on cardiovascular diseases and may lead to myocarditis or cardiac failure. The clove-like spike (S) protein of SARS-CoV-2 facilitates its transmission and pathogenesis. Cardiac mitochondria produce energy for key heart functions. We hypothesized that S1 would directly impair the functions of cardiomyocyte mitochondria, thus causing cardiac dysfunction. Methods: Through the Seahorse Mito Stress Test and real-time ATP rate assays, we explored the mitochondrial bioenergetics in human cardiomyocytes (AC16). The cells were treated without (control) or with S1 (1 nM) for 24, 48, and 72 h and we observed the mitochondrial morphology using transmission electron microscopy and confocal fluorescence microscopy. Western blotting, XRhod-1, and MitoSOX Red staining were performed to evaluate the expression of proteins related to energetic metabolism and relevant signaling cascades, mitochondrial Ca2+ levels, and ROS production. Results: The 24 h S1 treatment increased ATP production and mitochondrial respiration by increasing the expression of fatty-acid-transporting regulators and inducing more negative mitochondrial membrane potential (Δψm). The 72 h S1 treatment decreased mitochondrial respiration rates and Δψm, but increased levels of reactive oxygen species (ROS), mCa2+, and intracellular Ca2+. Electron microscopy revealed increased mitochondrial fragmentation/fission in AC16 cells treated for 72 h. The effects of S1 on ATP production were completely blocked by neutralizing ACE2 but not CD147 antibodies, and were partly attenuated by Mitotempo (1 µM).
Conclusion: S1 might impair mitochondrial function in human cardiomyocytes by altering Δψm, mCa2+ overload, ROS accumulation, and mitochondrial dynamics via ACE2.
FYI: The cell line used in this paper is a cardiac cell line, cardyomyocytes are the cells responsible for the contraction of the heart.
For a couple of years by now I have stated multiple times, from both observation and (lots) research that cardiac damage and inflammation had two main causes that cascaded into everything else, and arguably one of the main causes leads to the other, those being:
immune-mediated inflammation (aka infiltration)
mitochondrial dysfunction

Mitochondrial kinetics and function are nothing but complex, even though it is a very simple organelle (structures inside cells, often specialized). By using different methods to test mitochondria, from real-time testing to your “run of the mill” one’s authors found rather peculiar changes using just the S1 part of the Spike Protein.
Once again we are back at the S1 part of the Spike, arguably the most nocive part of the entire thing, here they found that in the presence of S1 alone within 24 hours the energy generation (ATP production) and mitochondrial respiration (using oxygen to convert nutrients into ATP) went up by using fatty-acids metabolism (breaking down fats to generate energy) leading to negative mitochondrial membrane potential, in simple terms means the process of energy stresses the powerhouse of the cell.
In case you forgot the S1 part is responsible for a lot of the clotting dynamics, proposed to be one of the inducers of Long Covid, bear mimicry with a bunch of other proteins, and is both parts of the Endotoxin-LPS mechanism, and the Galectin-3 fold.
At 72 hours in the presence of S1, respiration rate, and membrane potential decrease, while increasing ROS, mitochondrial calcium, and intracellular calcium. While all steps describe here and their harmony is important, this one is among the most important ones, if you are a long-time reader (and take some notes) you will remember me stating multiple times I don’t want free zinc, free iron, and free calcium around my body. Regulation of these nutrients is tightly controlled, and fluctuation of levels inside and outside the cells leads to long-term problems (and a hallmark of mitochondrial dysfunction).
Antibodies against ACE2 were able to stop this process entirely, but not antibodies against CD147, another important receptor that SARS-CoV-2 can use to hijack cells, and were attenuated by Mitotempo, a very specific mitochondrial antioxidant.
The conclusion speaks for itself, and as I wrote months ago, ACE2 is a master regulator of bioenergy inside us, and the virus messing too much with it (up and down) would cause mitochondrial problems.
The discussion section of this paper gives you a good overview of mitochondrial function and how bioenergetics occur inside the cell, it is complex but a good read for anyone interested.
Each part of the virus impacts mitochondrial function differently, but arguably the most important ones for our purposes are the S1/entire Spike and N (nucleocapsid) but other regions, conserved (unchanged) are also important long-term.
Given how connected the relationship with the Kynurenine Pathway and Mitochondrial function are, these dynamics and research are of high importance for the proper care and long-term outcomes for many, especially the vaccinated, which are put in a heavier oxidative (ROS) burden from the get-go, and each step of the way are bombarded with more and more bioenergetic disruption.
Far from me from completely generalizing within this specific subject (cardiac-related events from SARS-CoV-2 Spike protein, regardless of source) we are once again back at, if administration of antioxidants was proposed very early on, we could have mitigated a lot of the cardiac damage that occurred, and that will come.
Since I am not a big fan of being light on content, avoid watering down what I write (if I don’t push myself I see very little point in most things), and one of my traits is connecting things. The Kynurenine Pathway has a big footprint on cardiovascular diseases. (all the following little excerpts are from the paper I just linked), also, anything in parentheses is my own commentary/addition, unrelated to the paper itself.
These findings show that Ang II positively regulates kynurenine pathway. Also, kynurenine pathway activation in hypertension can be a sign of RAAS overactivity.
(dysregulation of ACE2, leading to an increase of Ang II which basically sets off inflammatory cascades inside you, the precise mechanism which I and Doorlesscarp both warned bivalent boosters would damage hearts even more).
Endothelial dysfunction upregulates IDO and increases kynurenine production. Kynurenine activates adenylate and soluble guanylate cyclase pathways and dilates arteries. 16 Endotoxemia enhanced endothelial IDO production in the mice model, thereby increasing kynurenine level and decreasing blood pressure.
Kynurenine is strongly produced in patients with idiopathic pulmonary arterial hypertension (PAH) and contributes to lower pulmonary arterial blood pressure by activating nitric oxide (NO)/cGMP and cAMP pathways in the pulmonary arteries.
These findings suggest that the kynurenine pathway is a compensatory mechanism that is activated in atherosclerosis and attempts to protect against atherosclerosis.
Besides, kynurenic acid, anthranilic acid, 3-hydroxykynurenine, and 3-hydroxyanthranilic acid impair myocardial mitochondrial respiration, which heavily disrupts cardiac energy and calcium homeostasis, increases ROS production, and contributes to the development of heart failure.109-111
Such as Galectin-3, Kynurenine, and its metabolites are also big double-edged swords, under very specific circumstances they can be protective, and under many others, they will often be damaging, with the only exception to this rule being the long-term presence or higher quantity of anything involved in the KP, which is a low-grade “bomb”, persistent low levels of inflammation, damage, and dysfunction.
Here is an example of Galectin-3 offering protection by inducing IDO1/Kynurenine pathway, protecting the kidney in this particular instance by the literal opposite pathway that incidentally the mRNA SARS-CoV-2 induces at really high levels.
Readers should always keep in the back of their minds the Galectin-3 dynamics of the Spike, the Kynurenine Pathway, and the Endotoxin interaction, they all share many aspects and are basically a singular, highly complex pathway that cascades into a lot of what we covered. With the role of endotoxins being heavily present in Covid cases, here is something interesting.
NAD+ ameliorates endotoxin-induced acute kidney injury in a sirtuin1–dependent manner via GSK-3β/Nrf2 signalling pathway
NAD+ was notably decreased and negatively correlated with kidney dysfunction in AKI, restoring NAD+ with NMN significantly ameliorates LPS-induced oxidative stress and apoptosis and attenuates renal damage. We also found that the protection of NAD+ is associated with SIRT1 expressions and performs in a SIRT1-dependent manner.
NAD+ (nicotinamide adenine dinucleotide) is a ubiquitous hydride acceptor that plays an essential role in energy metabolism and adaptive stress responses.8, 9 The kidney is both among the highest mitochondria and NAD+ containing organs, yet is also highly susceptible to NAD+ depletion.10, 11 Oxidative stresses and mitochondrial damage are drivers of sepsis-induced AKI, followed by cellular NAD+ depletion and NAD+ synthesis reduction.
And since we talked about Kynurenine.
Tryptophan catabolites, inflammation, and insulin resistance as determinants of chronic fatigue syndrome and affective symptoms in Long COVID
Background Critical COVID-19 disease is accompanied by depletion of plasma tryptophan (TRY) and increases in indoleamine-dioxygenase (IDO)-stimulated production of neuroactive tryptophan catabolites (TRYCATs), including kynurenine (KYN) and quinolinic acid. The TRYCAT pathway has not been studied extensively in association with the physiosomatic and affective symptoms of Long COVID.
Methods In the present study, we measured serum tryptophan (TRY), TRYCATs, insulin resistance (using the HOMA2-IR index), C-reactive protein (CRP), physiosomatic, depression and anxiety symptoms in 90 Long COVID patients, 3-10 months after remission of acute infection.
Results We were able to construct an endophenotypic class of severe Long COVID (22% of the patients) with very low TRY and oxygen saturation (SpO2, during acute infection), increased kynurenine, KYN/TRY ratio, CRP, and very high ratings on all symptom domains. One factor could be extracted from physiosomatic symptoms (including chronic fatigue-fibromyalgia), depression, and anxiety symptoms, indicating that all domains are manifestations of the common physio-affective phenome. Three Long COVID biomarkers (CRP, KYN/TRY, IR) explained around 40% of the variance in the physio-affective phenome. The latter and the KYN/TRY ratio were significantly predicted by peak body temperature (PBT) and lowered SpO2 during acute infection. One validated latent vector could be extracted from the three symptom domains and a composite based on CRP, KYN/TRY, IR (Long COVID), and PBT and SpO2 (acute COVID-19).
Conclusion The physio-affective phenome of Long COVID is a manifestation of inflammatory responses during acute and Long COVID and lowered plasma tryptophan and increased kynurenine may contribute to these effects.
In line with previous evidence, cited by the authors themselves within the paper, we now have further evidence of the Tryptophan dynamics and KP in Long Covid, and what one could call “the obvious” one among many of the long-term problem from covid, long or not, Insulin Resistance.
Here they found these 3 markers are good explainers for the physiological effects of Long Covid, proposed here being mediated by inflammatory responses. There are quite a few points I disagree with the authors in the discussion section of the paper itself, but their findings are more evidence for what I have been pointing out for a while.
Some of the effects and detriments of tryptophan and the KP are very contextual (you will see me stating this more and more), they argue a lot of the negative effects are self-limiting as an explanation for the lower levels of specific markers (IDO being one of them) in the blood of a lot of the Long Covid people in the study. I will make the case later on for the contextual nature of these “self-limiting” aspects of the KP.
Understanding the complex relationship of Tryptophan, KP, and its metabolic and immune-modulating effects are of high importance for, once again, the long-term health outcomes of millions of people, since this is tied primarily to the viral replication, something all current variants excel on and is inducing depressive/anxious behavior in a LOT of people, regardless if they are vaccinated or not (most of the unvaccinated rebound fast, but these psychological changes are so drastic many are aware and uncomfortable with them).
I have a few other things to write about, but they don’t fit the “theme” here, so perhaps a shorter e-mail tomorrow, or longer who knows where research may go.
Wish everyone a great week ahead.
I am thankful for the subscribers who choose to support this work, and for people who use Kofi as a one-time thing.
Good for you. Taking time away is vitally important. We all need our refuges. Especially when you are dealing with such important information. Deep dives into the physiology of all this much appreciated. The heart of the matter.
One for you John Paul;
https://www.biorxiv.org/content/10.1101/2023.03.14.532609v1
Epithelial galectin-3 induces mitochondrial complex inhibition and cell cycle arrest of CD8+ T Cells in severe/critical ill COVID-19