


Spike Protein as a endotoxin delivery system
Part I

A warning of some sort, unlike the structure of most of my posts, which is basically just science, here there will be more opinions based on a couple of years of observation, and quotes from the paper will be in quotation marks and not quotation blocks.
The following post is almost required reading, even though I will explain myself and further elaborate on some of the points, you should still read it, it is a very complex topic. My most recent one on the Kynurenine Pathway, ME/CFS and Long Covid is a recommendation but not “required”, but at least check out the closing images and papers mentioned.
First, what is LPS.
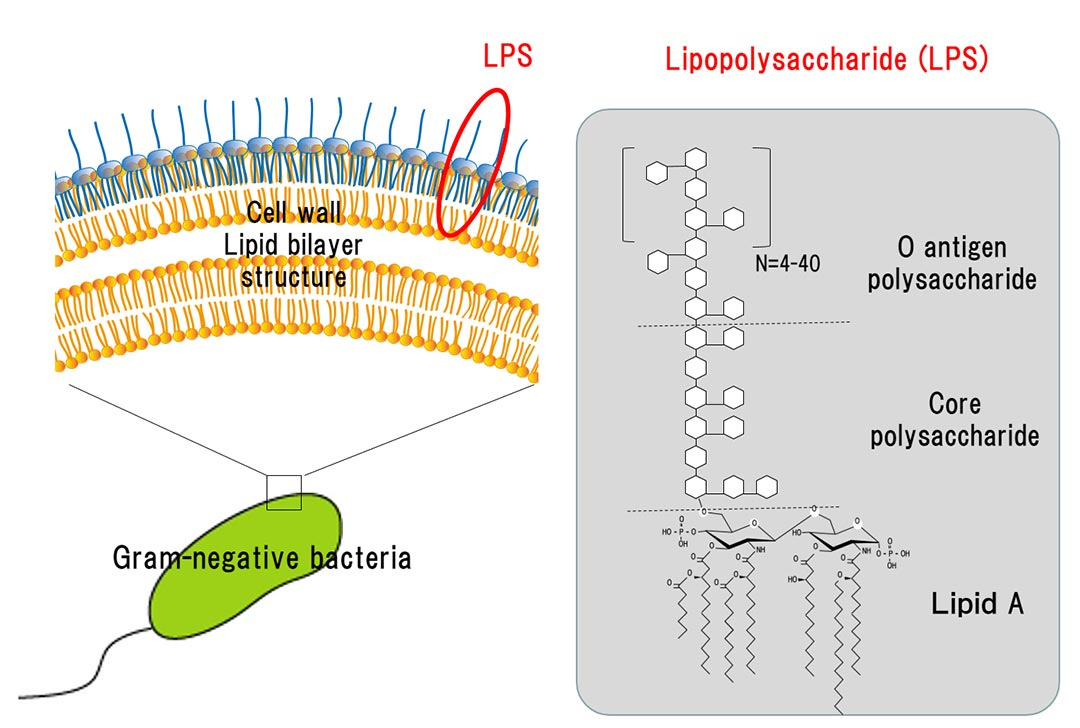
It is the outer part of the membrane of gram-negative bacteria. To give you an idea of how potent LPSs are to activate the immune system, you need 1.000 to 10.000x more Beta-Glucan and peptidoglycans for immune activation than you need LPS. It is one of the most potent “toxins” known to science.

In the paper covered above, the authors of following paper demonstrated that the Spike Protein interacted with LPS to form aggregates of Spike Protein, among other possible explanations. This one is very different, and it was finally answered by 14-month-old question.
Could the Spike Protein interact and bind to LPS to create endotoxic reactions inside the body ?
SARS-CoV-2 spike protein as a bacterial lipopolysaccharide delivery system in an overzealous inflammatory cascade
Patients with metabolic syndrome are at a higher risk to develop severe COVID-19 disease involving sepsis and ARDS). Interestingly, metabolic syndrome is associated with a high blood level of bacterial lipopolysaccharide (LPS) due to gut dysbiosis and translocation of bacterial components into the systemic circulation.
Non-survivor COVID-19 patients also presented increased systemic levels of LPS and soluble CD14 during hospitalisation compared to survivors, indicating a link between elevated LPS level and death.
A common “denominator” among people under dysfunctional metabolic states is the different degrees of gut dysbiosis (the disequilibrium between good and bad microbes in your gut), in fact, and as I covered here many times, there is extensive research on how insulin resistant states add fuel to the fire, creating what is called gut-derived endotoxemia. A hyperglycemic or dysfunctional metabolic state will contribute to poor immune function and cell sensing and raise the levels of bacteria and LPS inside the blood.
While using different pathways, a lot of the recent deaths directly from vaccination (as per the pathologists analysis) and from the infection since early 2020 have something in common. Bacterial overgrowth, excessive presence of bacteria in the blood. This will be rather important later on here.
The authors of the paper went on to further uncover and categorize if LPS and Lipid A (a part of LPS) could bind to SARS-Cov-2 S1 and S2. Using different methods to measure the shape, weight, and expression of the proteins they confirmed the aforementioned. LPS binds to both parts of the Spike, but there was more pronounced of said binding in the S2 part of Spike Protein, and the same was true for the Lipid A part of the LPS, and once again S2 had more affinity to “gluing” this than S1. “Futherrmore, in the presence of LPS, almost no visible S2 protein was observed, even at lower concentrations (e.g. 100 µg/ml), suggesting a stronger binding of LPS to S2 in its pre-fusion conformation”, in simple words, the S2 part of the Spike has a stronger binding of LPS before the Spike binds to a receptor to enter a cell.

The following section of the paper is fascinating on its own. Knowing where LPS and Lipid A bind holds much importance as uncovering the fact that they just bind to any subunit of the Spike. Each binding site has a different affinity for LPS binding. “This indicates that LPS binding to S1 observed in our BN-PAGE experiment does not involve the S2 pocket, but rather involves the pockets in the NTD and RBD only.” To bind to the S1 part of the Spike Protein it involved both the NTD and RBD. The NTD will be of particular importance later here.
The way Lipid A binds to the RBD resulted in said lipids being less exposed to solved compared to the binding of the NTD (different shapes) suggesting a higher affinity for that part of the Spike, there is also the difference in what each part can accommodate, the NTD can accommodate just small parts of the Lipid A section, while the RBD can accommodate the entire tail.
Due to the large size of the S2 pocket and the partially exposed lipid tails of bound LPS, we hypothesized that more than one LPS molecule could occupy the binding site through lipid–lipid interactions, which would explain the marked changes in migration pattern of S2 in the presence of LPS
The S2 pocket could therefore act as a pool for small LPS aggregates and may be able to serve a role in forward transfer along the TLR4 relay.
This is one of the most important sections of the entire paper, due to how “large” microscopically speaking the S2 pocket is, they hypothesize that it can have more than one LPS molecule at the binding site, which is very meaningful for the mechanisms and physiological effects we are about to discuss (and the reason I am not overcomplicating this analysis so far). The way your body responds to LPS is very much “shape and position” oriented, meaning the shape the molecules/parts of the LPS have and where they find themselves. With more LPS molecules in a protein, therefore a stronger effect, depending on which receptor it binds and activates.
LPS binding to S protein subunits boosts proinflammatory responses
Correspondingly, the prefusion stabilized S2 construct showed less intrinsic proinflammatory activity per se but retained a significant ability to boost the responses to ultra-low levels of LPS. Hence, the stabilized S2 retains the inflammation boosting activity previously demonstrated for the whole S protein.
S1 alone did not yield any measurable NF-κB activation. S1 combined with 2 µg LPS resulted in a significant proinflammatory response. S2 alone yielded a proinflammatory response per se, which was not unexpected given the detected LPS content in this preparation.
Taken together, the results showed that S1 has a boosting effect on LPS induced inflammation. Indirectly, the results also showed that S2’s inherent proinflammatory effect was due to minute LPS-contaminants, resulting in a boosted NF-κB response to the combination. The proline-stabilized S2 construct, which contained less contaminating LPS, exhibited a low proinflammatory activity alone, but significantly boosted the response to 2 µg LPS. Intriguingly, S1 displays a boosting effect on LPS-induced inflammation in vivo but not in vitro, suggesting additional mechanisms in the former. We hypothesize that one potential reason for this in vitro–in vivo difference is the interactions with proteases secreted in the tissue during inflammation, such as neutrophil elastase (NE) that could modify the structure of the S1 subunit and alter its affinity to LPS.
This supports our hypothesis that LPS binding to the high-affinity sites in S1 in vivo is compromised by proteolysis during inflammation resulting in an onward transfer to LPS receptors. The proteolytic sites of these enzymes and how they weaken LPS binding, however, would require further studies.
The S2 itself has the ability to boost the inflammatory response elicited by LPS, even at very low quantities of LPS. And so did S1 possessing a boosting effect on the inflammatory response induced by LPS, and this S1 boosting ability is present in vivo but not in vitro, because of the inflammatory response present in vivo, and therefore the protein breakdown from the inflammatory response.
Therefore the binding into S1 will be affected in vivo during inflammation.

By testing for contamination of commercially available Spike protein preparation, finding the exact levels of said contamination, and thoroughly testing to exclude it from the results, the authors elegantly proved that the Spike Protein is an intermediary, a delivery system for LPS, which in turn sets off the hyperinflammatory response, rather than the Spike Protein being the causative agent.
LPS binding to the NTD and RBD is interesting due to their instrumental role in immune evasion of SARS-CoV-2. Haem metabolites binding to the NTD yields profound conformational changes that inhibit neutralizing antibodies (Rosa et al., 2021). As LPS binds to the same pocket, it is possible that it could also alter the conformation of the antigenic supersite (McCallum et al., 2021b). Linoleic acid binding to the RBD bridges two adjacent RBDs (Toelzer et al., 2020), hence favouring the down state, which reduces accessibility to antibody epitopes. Similarly, the charged headgroup of LPS protrudes out of the binding pocket and could potentially interact with a neighbouring RBD and shift the open–closed equilibrium of the S protein.
Not only the LPS can bind at different sites (parts) of the entire Spike Protein, but in certain sites, it will most likely alter the conformation (shape) of different parts, in this case, it could alter the conformation of the antigenic supersite, which is another mechanism for antibody evasion (or the opposite, antibody attraction).

Now we delve into the conclusion of the paper, one by which I was surprised, as you can see, the LPS binding sites do not make sense from a structural standpoint, they offer little evolutionary pressure but the authors, say, without actually saying that this might be a byproduct of the famous insertion of unnatural sites.
The second highlight is just the cream on top. Since I have argued for over 2 years that the virus and the Spike Protein interact with bacteria, and have bacteriophage-like properties or behavior. It remains to be proven, but I stand firm by that old assertion, as most of my observations were proved to be correct via multiple mechanisms later on. As a friend recently wrote to me, when you are targeting or engineering something, there are a lot of unknown effects, and not everything was created with a purpose. So this interaction most likely was a fluke.

Synthetic biology biggest fluke in human history. To summarize, what we just covered here demonstrates that the Spike protein is a delivery system for LPS, an endotoxin, which is the de facto mechanism for the potent TLR4-mediated inflammatory response and damage we see around Covid. And multiple regions of the Spike help this mechanism take effect. Here is where the papers end, and where we begin.
If you read my other LPS Substack, you will read my opinions and observations I made over a year ago. One of the said assertions was “Could the Spike Protein interact with bacteria or LPS and modulate the inflammatory responses and all signaling and cascades ?” , in that moment I thought it was merely me thinking the way I do, adopting a complexity perspective, looking at everything from a non-linear dynamic.

Given what we just covered so far, it is now not only possible, but plausible taking into consideration other effects of the Spike Protein (it works as a membrane piercer) that the Spike Protein interacts, piercer, deconstructs bacterias and biofilms, and adds it to its own structure. This is where things get a little complicated.
One of the most remarkable aspects of SARS-CoV-2 is its inflammatory signaling, its rewiring of cells into an inflammatory state, and its potent activation of inflammasomes. Everything occurring so far was outside the cell, the following occurs inside the cell, and has enormous implications.
Inside the cytosol (the liquid inside the cell) there are enzymes called Caspases and they are responsible for inflammation and cell death, especially pyroptosis, they will respond to intracellular pathogens, altered molecules (LPS) or intracellular structures. Inside the cytosol these Caspases in two ways, a canonical way, and a non-canonical way. The non-canonical response hinges more on the shape and molecular aggregates of LPS inside the cell different structures of the LPS will determine the possible receptors the cell uses to recognize LPS, in this sense they are not merely enzymes but “Pattern recognition” sensors. The Caspases activated in this manner are 4 and 11.
Galectin-3 promotes noncanonical inflammasome activation through intracellular binding to lipopolysaccharide glycans

The importance of the finding above can not be understated. My next post will be the Galectin-3 one, but if you are a new reader or not aware, there is a substantial presence of Galectin-3 in many disease states, especially present in hyperglycemic metabolic states, but also in SARS-CoV-2 infection, and a portion of the NTD is literally a mimic of Galectin-3 therefore inducing the expression (making the body produce more) of it.
Inside the cytosol, in the presence of glycans (small sugar “parts”) Galectin-3 will bind to LPSs and amplify the Caspase-4 and 11 activation causing more pyroptosis and more inflammation. This is of importance because of the following.
Caspase-4/11 exacerbates disease severity in SARS–CoV-2 infection by promoting inflammation and immunothrombosis

The authors of the paper above were not aware, by any stretch of the imagination, of this most recent paper on Spike-LPS interaction, but their findings were substantial at the time, and in light of this new mechanism, even more impactful. In regards to severity of disease, the Spike-LPS endotoxic behavior is one of the main drivers of inflammation, thrombosis, endothelial damage, producing severely more strong responses when the mechanism is initiated inside the cell (cytosol) through Caspase 4 and 11.
Caspase 4 has other interactions with LPS, being able to dissagragate to smaller structures and forming small complexes being one of major interest.
Inside the cell it will interact and bind with Galectin-3, further driving inflammatory damage, inflammatory cell death (pyroptosis), all sorts of dysfunction and severity of disease. In fact, Galectin-3 has been proposed as a new marker for potential prognostic for disease severity in SARS-CoV-2 infected patients. The presence of LPS in the blood stream also activates the Kynurenine Pathway, it explain the gut issues, dysbiosis, bacterial translocation (bacteria leaving certain regions of the body), the mechanism described here explain a sizable portion of everything going on.
And now I want you to remember we inject billions of people with the Spike Protein, and nothing they supposedly did, changed the capacity of the Spike Protein to do any of this.
I will inevitably write a Part II soon, but more exploratory.
Thank you and big appreciation for all supporters here and on KoFi, and also everyone who shares, also helpful !
Keep in mind that this experiment (covid) was conducted on a population that is widely vitamin D deficient. Against that backdrop, the final common path of covid and covid vax injuries is harmful inflammation exacerbated by dysfunctional vitamin D deficient immune systems. It’s nice to know how the spike protein is killing us but more important to know how to prevent it…take your vitamin D.
Does this suggest a need to add some dysbiosis mitigation supps to the stack? Probiotics, tryptophan, colustrum right when infected or after jabbed to prevent escalation?