
Immune dysfunction and immune "aging" up to 3 years after Covid

Consider becoming a paid subscriber, and if you are, thank you very much !

A few weeks ago, I wrote the following article, belonging to an overall theme and research I have done over the years, and in my opinion, critical to understanding both the viral damage, immune dysfunction, and also mRNA dysfunction, damage, etc.

We finally have a long-term follow-up study that maps the highly complex immunological shift in Long Covid patients. This shift appears to be a shared pathway across all degrees of infection, with the distinction between groups being how long they remain in this dysfunctional state. I must preface this by noting that this study was conducted by Chinese researchers in China.
T cell-driven sustained inflammation and immune dysregulation mimicking immunosenescence for up to three years post-COVID-19
Discrepancies between different studies on Long Covid are complex, but one reason is simple: a lack of long-term follow-up. A 12-month follow-up is already challenging, but a multi-year follow-up with multiple testing rounds is significantly harder. All tests are snapshots in time. Thankfully, the authors of this study took a long-term approach, conducting comprehensive immune profiling of 47 patients over 3 years. The cohort included 24 moderate, 14 severe, and 9 critical cases. The timeline was as follows:
Hospitalization in March 2020 (HO) → Follow-up in July 2020 (F1) → Second follow-up in December 2020 to January 2021 (F2) → Third follow-up November to December 2021 (F3) → Follow-up March to April 2023 (F4)
A total of 25 healthy controls (HC) were used, 7 recruited for the study, and 18 from published datasets.
Over the 3 years, NK cells, conventional dendritic cells, and megakaryocytes showed progressive recovery, while certain T-cell subsets, monocytes, and plasma cells had abnormalities even at the 3-year mark. Cytokines (inflammation) were distinct over the 3 years. Remarkably, the levels of IFN-γ, IL-1β, IL-17, IL-4, IL-9 and MIP-1b remained elevated in convalescent patients compared to healthy controls even three years after infection. Most of my reader memories and synapses will instantly flare up after reading the bold text.
Monocytes, which could be called the primary line of defense of our immune system, are known to undergo dysfunction after an infection and drive early inflammation,. Their inflammatory signaling can be observed at the 1-year mark, dominated by the markers CCL3 and S100A (an alarmin that can activate TLR4 and RAGE pathways).
After the first year, inflammation, which was previously thought to be driven by non-classical monocytes, but here at year 2 and 3, most monocyte subsets stabilize toward normal levels, yet systemic inflammation persists. Thus, they deduce it is not driven by monocytes beyond the first year.
Specifically, pro-inflammatory classical monocytes (high in CCL3 and S100A8) rapidly declined after acute infection. Meanwhile, another classical subset (characterized by high CCL3 but lower S100A) increased, and they proposed that they may exert anti-inflammatory effects. The non-classical monocytes (CDKN1C⁺), which drive inflammation at the one-year mark and correlate with symptoms, trended toward recovery over years 2 and 3,[though remaining slightly altered. Since monocytes normalize after 3 years, what then drives the persistent inflammation?
Looking into the source of continued inflammation, and in line with many papers I covered this year, there was a decrease in naive (new) CD4, CD8, and MAIT cells. MAIT cells are Mucosal-Associated Invariant T-cells, critical cells against bacterial and viral infections, and per their name, they are abundant in mucosal tissues like the gut. A decline of CD4 and CD8 is associated with the aging of the immune system. A subset of CD8 T cells (marker ZNF683) remained elevated throughout the 3 years.
This is an important marker because ZNF683 is a marker for effector, short-lived cells. But here, there is a chronic elevation, resembling memory-like cells that are chronically inflammatory. This indicates there is either persistent antigenic stimulation (Spike, viral fragments, RNA complexes) or bystander activation (other cells maintaining signaling).
Over time, the levels of Th17 cells and their subsets were consistently higher than healthy controls, and so was their main marker, IL-17, present in high levels even after infection. But the two subsets of Th17 cells had distinct immunological effects. LTB+ Th17 cells (Lymphotoxin Beta is a key cytokine for regulating the immune system) were elevated at hospitalization, declined at the first visit, but persisted at almost twice the levels found in healthy controls until the fourth visit.
Their relationship with inflammation was opposite. LTB⁺ Th17 abundance was positively correlated with inflammatory cytokines (IL-4, IL-9, IL-17, IL-1β, IFN-γ, MIP-1b), while RORC⁺ Th17 cells were negatively correlated. Gene expression analysis confirmed this dichotomy, RORC⁺ cells expressed high levels of genes involved in cell migration and regulation (S100A4, S100A10, S100A11, LGALS3, ANXA2), while LTB⁺ cells expressed genes related to general immune activation and Type I interferon responses.
LTB+ Th17 cells in the blood were positively correlated with several Long Covid symptoms, such as anxiety, depression, chest pain, higher pain sensitivity, and smell disorders. The opposite was true for RORC+ Th17 cells. Further analyses to explore the molecular drivers of both Th17 subsets revealed distinct clusters.
LTB+ → PTPRC, SIRPA, S100A8, EBI3, LAMA5, IL16, TGFBI and CLCF1
RORC+ → JAM3, CCL3, NAALADL1, COL18A1
IL-16 and S100A8 are highly important here because the former acts as a danger signal to everything around its origin, and as previously described, it can create feedback loops with important inflammatory receptors (TLR4 and RAGE). IL-16 attracts white blood cells and is inflammatory, acting on CD4 cells (which are the origin for Th17). Both IL-16 and S100A8 effects were validated by stimulating the blood cells of the patients with them, resulting in the increase of RORC+ Th17 cells, thus validating their role in inducing LTB+ cells.
What is happening (or hidden in complexity)
Some of the markers and genes here are highly significant and help us get a broader picture, integrating this paper with many others. RORC+ cells here express LGALS3, which is the gene responsible for… Galectin-3, which plays a paradoxical role, it can both induce chronic inflammation and create a complex cascade of effects, but it can also “tackle” these responses, because, as I previously mentioned, Galectin-3 can negatively modulate Th17 responses.
Another hint was the MAIT cells, which suffer severity-dependent depletion and do not recover over time. MAIT cells are very important for mucosal defense, but borderline critical as antibacterial defense. This reduction leaves someone predisposed to secondary infections.
S100A8 itself offers another clue. They remained significantly high even 3 years, and while they are initially derived from monocytes, these undergo normalization so there must be alternative sources. Epithelial cells that are damaged, platelets, and especially NETs, Neutrophil Extracellular Traps, S100A8 is a coro NET component, forming micro structures that resist degradation and could explain persistent high levels.
IL-17 by itself damages epithelial barriers, and in specific conditions, it can damage the gut barrier, causing small amounts of endotoxin to leak into the blood, this can be observed and inferred by the markers IL-1β, and MIP-1β present here. Chronic exposure or high levels of IFN-γ and TNF-α will further compromise barrier integrity.
This creates a core feature and an old problem in regards to SARS-CoV-2, Spike exposure, and Long Covid. Damage to cells, tissue, or exposure to endotoxin, even at low levels, will make the body produce HMGB1, which amplifies all inflammatory signals here. In the gut, it will increase MLCK via RAGE, which further compromises bacterial translocation. HMGB1 can force systemic metabolic shift and induce the Warburg Effect, and Th17 are THE MOST sugar hungry immune cells in the body.
The sustained expression of S100A8 amplifies HMGB1 release on its own, as NETs formation will make the neutrophils release HMGB1, which increases production of S100A8, creating a self-sustaining feedback loop of inflammation. The chronic activation of these pathways and specific receptors, such as TLR4, RAGE creates a paradoxical state of tolerance while specific inflammatory signaling remains very active. For the visual learners.
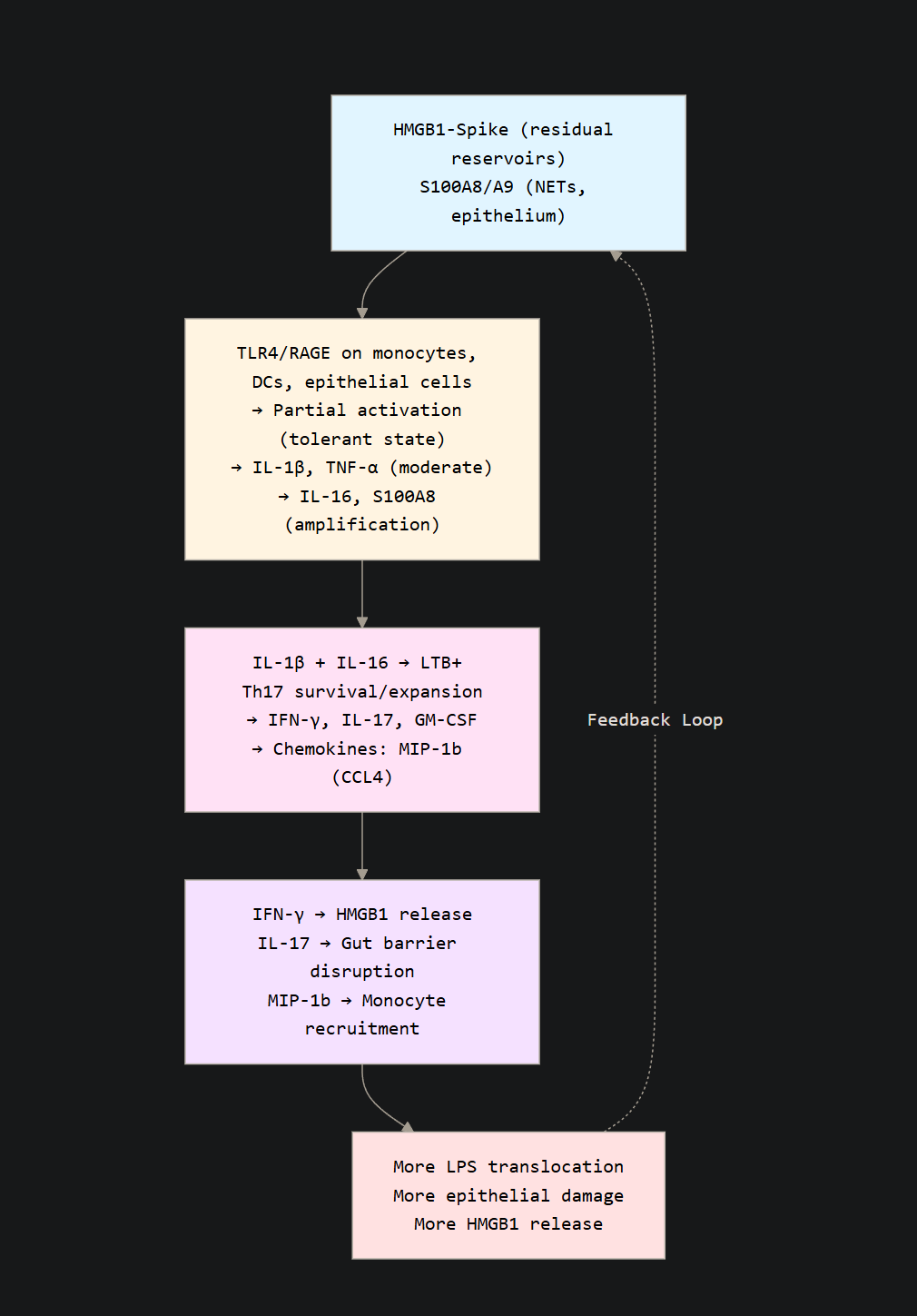
Lastly, the paper itself demonstrates something I have been concerned about and hinting at for months. Thymic damage, which is not a proposition, but a verifiable, measured, and published result of infection. The naive T Cells loss indicates lower thymic output, while the other signals push for differentiation, and HMGB1 by itself induces this. The thymus has its specialized epithelial cells. Two proteins are critical in the thymus.
IL-7. IL-15. And this will be covered in the next article. And merely to add to my recent article on Metformin, this study on the effects of Metformin taken shortly after infection and Long Covid. Not only does Metformin lower your chance of developing Long Covid even when taken shortly after infection, but it is one of the few drugs that can help the thymus. It is a great option for those who may need it, healthy people don’t necessarily need it.


I am already working on the next few articles. Hopefully, I will publish in the next few days before my birthday (December 9).
For the most part, I usually try to be strictly good vibes during December, holiday season and all, but given how rough this past quarter was for me, I am behind on a lot of writing. Thus, I must research, write, and publish.
I wish you all a good week ahead.
Thank you! As metformin seems to be promising in a lot of things from arthritis to alzheimers, the insulin metabolism seems to have a really big role in health&sickmess. So when looking at that, I bumped into brain insulin resistance. Possibly existing without obesity/diabetes. And as ketogenic diets help epileptics, the brain insulin signaling, if messed up, can cause serious trouble. If brain has ended up insulin resistant, the only way the brain gan get adequate energy is from ketones. So, mct oil, caprylic acid, c-8, whatever you call it, may be good, even without ketogenic diet.