
Epstein Barr link to Multisystem Inflammatory Syndrome in Children
Paradoxical Immunosuppression

As someone deeply afflicted by Rubik’s complex (the irresistible desire to solve puzzles, especially complex, layered ones), Multisystem Inflammatory Syndrome (MIS) has been an interest of mine ever since it surfaced, which occurred at first in Children.
Aggressive, systemic inflammatory conditions are usually highly complex and verge on paradoxical territory more often than not, such as early Covid “cytokine storm”, and MIS-C. Among my explorations in MIS, I have come to believe that Herpes Viruses would be central players in this delayed, systemic inflammatory reaction.



AI-generated podcast, with a massive caveat. Given the sheer complexity of each paper referred to in the articles above, let alone this one, the AI loses significant nuance; thus, the audio is merely an overview.
TGFβ links EBV to multisystem inflammatory syndrome in children
The authors open by mentioning and covering other research, each covering an aspect of MIS-C, such as how a group of patients (from another research group) had IL-1Ra-neutralizing autoantibodies (the body attacks those proteins), which in that setting helps explain the massive inflammation. Another citation is the superantigen-like nature of the Spike, and the expansion of a very specific T cell subtype, named TCRVβ21.3+ (this will be very important here), although the authors cite this paper which the authors found MIS-C in SARS-CoV-2 negative children, pre-pandemic, the argument meaning it isn’t necessarily mediated by the Spike Protein.
The authors used a multi-centre approach, meaning using 6 different places (in 4 different continents), with 145 MIS-C patients, and 221 controls.
TGFβ is uniformly upregulated in MIS-C
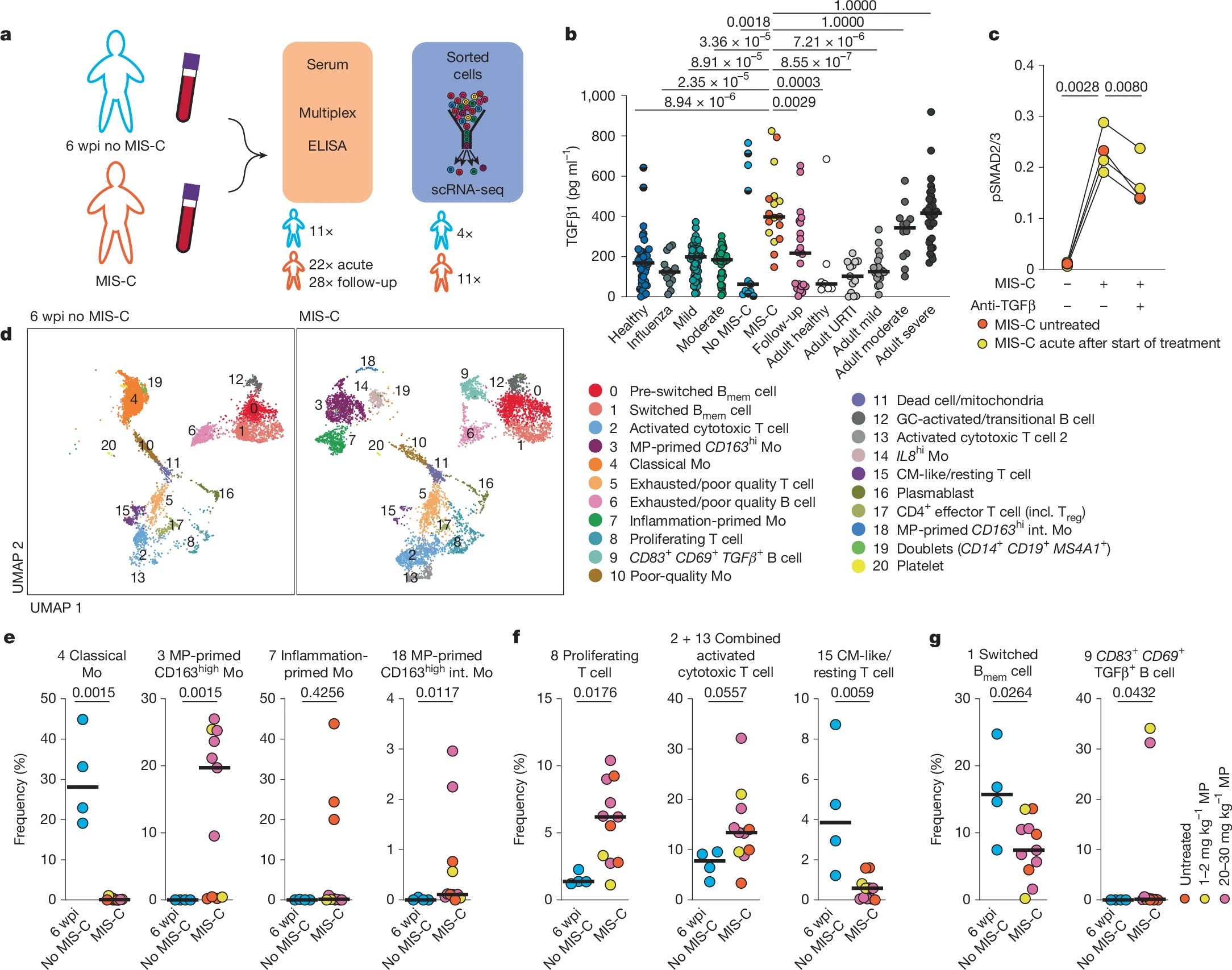
To get a more comprehensive picture of this complex condition, the researchers measured a wide range of cytokines and chemokines in patients during the acute phase of MIS-C, as well as at follow-up visits. They used children with SARS-CoV-2 infection at 6 weeks post-infection, but without MIS-C, as an at-risk control group. They observed upregulation of Type 1, Type 2, and Type 3 interferons (keep Type 2 in mind, specifically IFN-gamma, as it will be important later).
TGFβ1 is a potent modulator of the immune system, often acting as a “master” anti-inflammatory, dampening over-the-top immune reactions. It's known to be elevated in severe COVID-19, and this study confirms that in MIS-C as well. MIS-C patients had TGFβ1 levels comparable to severe COVID-19 cases, and almost 3 times higher than non-infected children, and 7 times higher compared to children infected with SARS-CoV-2 but without MIS-C. Importantly, TGFβ1 levels decreased after treatment with immunoglobulin or methylprednisolone, therapies known to reduce inflammation in MIS-C.
The author’s add to the complexity by citing other evidence, such loss of the gut mucosa in certain MIS-C patients is linked to TGFβ regulation, and perhaps viral persistence in the gut or fragment persistence in my assessment, the Nucleoprotein of the virus interacts directly with SMAD3 in turn activating theTGFβ pathway and this creates a feedback look. The Spike Protein itself interacts with integrins and activates latent TGFβ. In their words, “Increased TGFβ may result from an excessive TGFβ response during acute SARS-CoV-2 infection in these children or from viral persistence”.
TGFβ depends on being “cut” to act in the cell, high levels don’t reflect biological function, so the authors used blood from MIS-C and incubated it with T cells from healthy donors, and this in turn increased phosphorylation of SMAD2/3. This is a necessary step for TGFβ activity.
Through a painstaking process of single-cell RNA sequencing and detailed analysis of various immune markers, the authors found that TGFβ leaves a distinct "imprint" on MIS-C T-cells. Specifically, they observed activated and cytotoxic T Cells being particularly prominent, alongside a loss of memory B cell class switching.
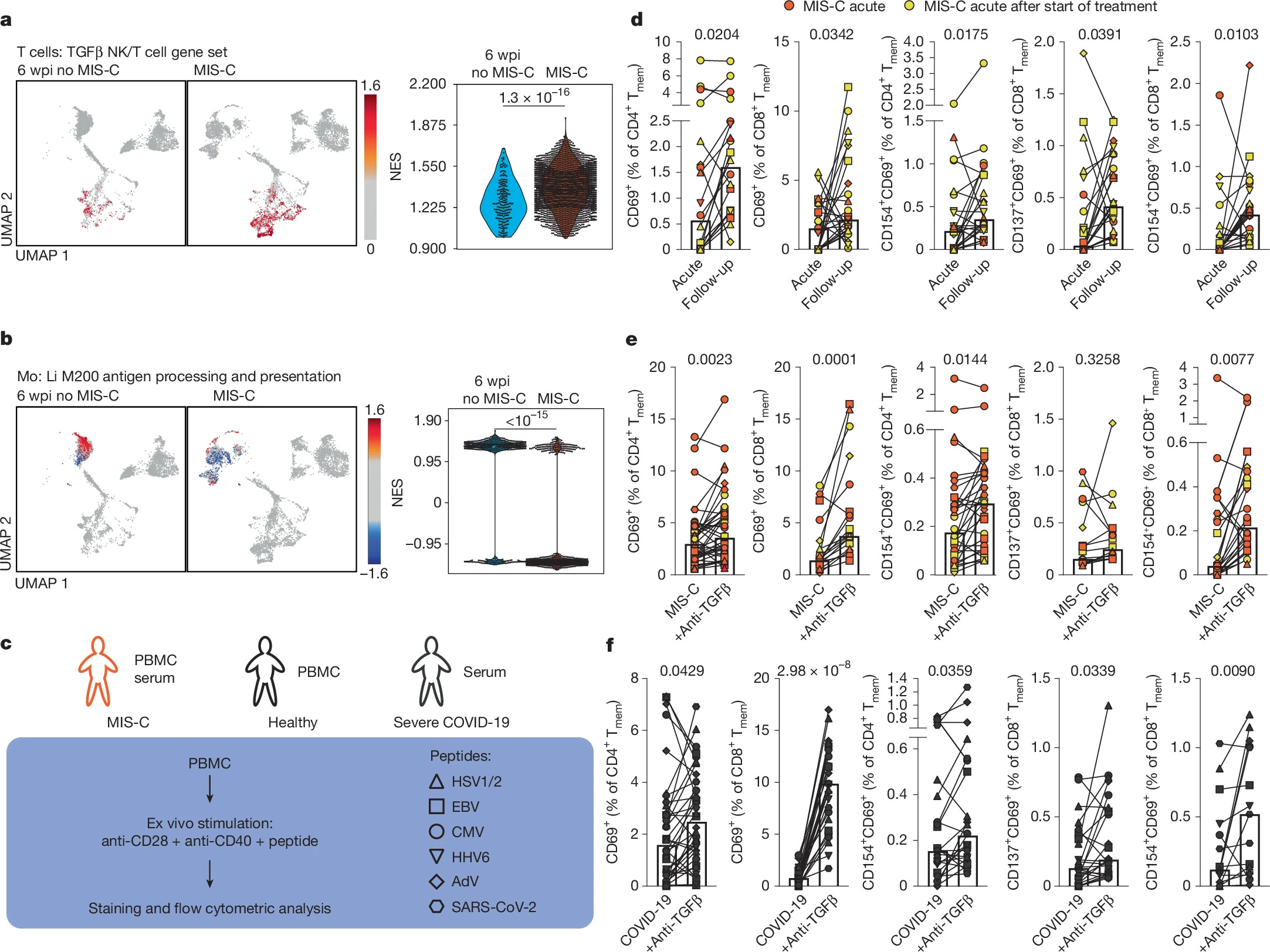
If there is one graph of this paper you should analyze, and perhaps internalize is the following one, especially figures D and E (all the small symbols and lines). Both panels show a general memory T-cell impairment in MIS-C when exposed to peptides from, mostly, Herpesviruses, and this is mediated by TGFβ, as using anti-TGFβ treatment reverses the impairment. Meaning the T-cells can’t quite fight off these viruses.
TCRVβ21.3+ T cells react to EBV peptides
MIS-C is associated with expansion of CD4+ and CD8+ T cells with TCRVβ21.3, but this expansion is not universal, it has specific variation, and it is test dependent. For future reference, whenever you see TCRV-add classifier here- it means these are contextually specific, and this is the case, and the authors sought to understand this specificity.
By comparing the T-Cell Receptor atlas (long list) with the TCR of MIS-C, the authors found a significant overlap between MIS-C TCR and EBV-specific TCR, with β21.3, T cells being clustered with EBV-specific TCR from multiple donors, suggesting these cells might be EBV specific.
To directly test this EBV specificity, the researchers exposed T-Cells to EBV-derived peptides (small protein fragments), particularly focusing on the EBNA2 protein. EBNA2 is a key protein expressed by EBV during lytic reactivation (when the virus "reactivates" inside a latently infected cell, begins to replicate, eventually killing the cell and spreading around). They found that healthy donor T-Cells exposed to EBNA2 peptides showed a consistent enrichment of TCRVβ21.3+, confirming that these T-cells are indeed capable of recognizing "parts" of EBV, specifically EBNA2.
They isolated and enriched TCRVβ21.3+ T cells from healthy donors and co-cultured them with autologous EBV-transformed B cells (lymphoblastoid cell lines, LCLs). TCRVβ21.3+ CD8+ T cells exhibited increased killing capacity against EBV-infected B cells compared to control T cells depleted of TCRVβ21.3. These are functional EBV-specific cytotoxic T cells.
They then investigated whether TGFβ's immunosuppressive effects impacted the ability of these EBV-specific TCRVβ21.3+ T cells to control EBV-infected cells in MIS-C. They found that despite the expansion of TCRVβ21.3+ T cells exhibited impaired cytotoxicity and killing capacity in MIS-C patients.
These T cells are incapable of efficiently attaching themselves to EBV-infected cells, in a similar fashion to TGFβ-affected NK cells that do not attach to SARS-CoV-2-infected cells. TGFβ overproduction is also used as an immune evasion mechanism. TGFβ alone can cause EBV reactivation, thus, the authors tested if its prolonged presence could induce it, and found that yes, by suppressing T-Cell’s ability to kill infected B-Cells, it facilitates reactivation.
To confirm the prevalence of EBV reactivation, they measured EBV seroprevalence in MIS-C patients and control groups. Strikingly, MIS-C patients exhibited significantly higher EBV seroprevalence compared to age-matched healthy children and children at risk for MIS-C post-SARS-CoV-2 infection. Furthermore, they detected EBV mRNA transcripts in single-cell transcriptomes of B cells and plasmablasts from MIS-C patients, and EBV DNA in cell-free plasma from a subset of MIS-C patients, providing direct evidence of EBV reactivation in vivo.

If you took note of Type II Interferon, and read or listened to the other articles, you will know a hallmark of MIS-C, in similar fashion to all the paradoxical effects of SARS-CoV-2, is the high expression of Interferon-Gamma (IFN-y). The MIS-C papers covered here all observed elevated levels of IFN-y, here less abundant than others, but still present.
Both TGFβ and IFN-γ can stimulate BAFF expression (BAFF is a potent B-Cell activator). At high enough levels, this will cause an imbalance not only in the proteins regulating B-Cell function, but also “tilt” the body into autoreactivity, meaning the process of developing autoimmune diseases. BAFF also affects the function of many helper cells. Under specific circumstances, it delays and exaggerates T-cell immune responses, called “Delayed-type Hypersensitivity”. A delayed hyperinflammatory response is the hallmark of MIS-C.
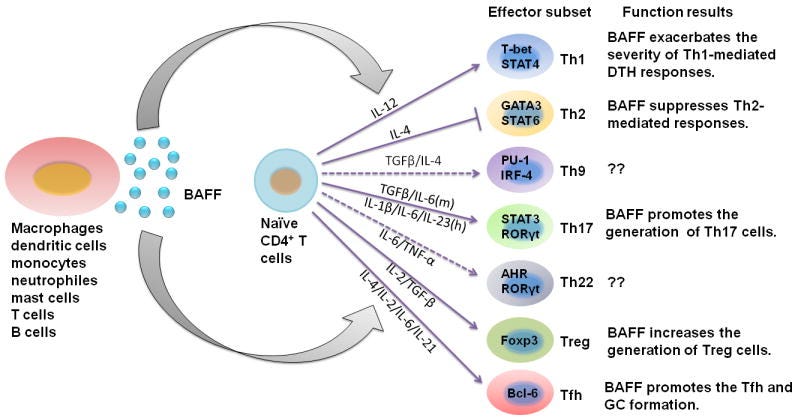
TGFβ has a paradoxical relationship with IFN-γ, where the first is produced to control the strong inflammatory response of the second, and this is even more evident in relation to SARS-CoV-2 if we take into account the Spike Protein-Endotoxin behavior, which I named “Neo-polymorphic Toxin”. This neopolymorphic toxin complex binds to and activates and amplifies TLR4, which alone can cause hyperinflammatory, or sensitizes the body to a subsequent inflammatory trigger, leaving the body primed for a secondary hit, this time delayed.
SARS-CoV-2 exposure as an initial trigger, Spike Protein+LPS complex
Significant production of inflammatory proteins such as IFN-y, IL-6, TNF-a, and other inflammatory mediators and inflammatory proteins
TGFβ production and release as an attempt to regulate this strong inflammatory response, or by tissue damage and cellular stress
Elevated TGFβ impairs T-Cell reactivity and cytotoxicity, affecting Herpesviruses, here specifically EBV
EBV reactivates from B cells, which in turn will contribute to HERV-K18 expression, among other dysregulations
Antigens usually persist for days, sometimes weeks (respiratory viruses in particular), and this secondary hit may arise from the delayed manner in which the body clears viral remnants. The secondary hit may also arise from a subclinical or transient exposure to superantigens, not necessarily Spike Protein-derived, but from other pathogens, such as Staphylococcus aureus, or Epstein-Barr reactivation leading to the expression of HERV-K18, thus creating a superantigen.
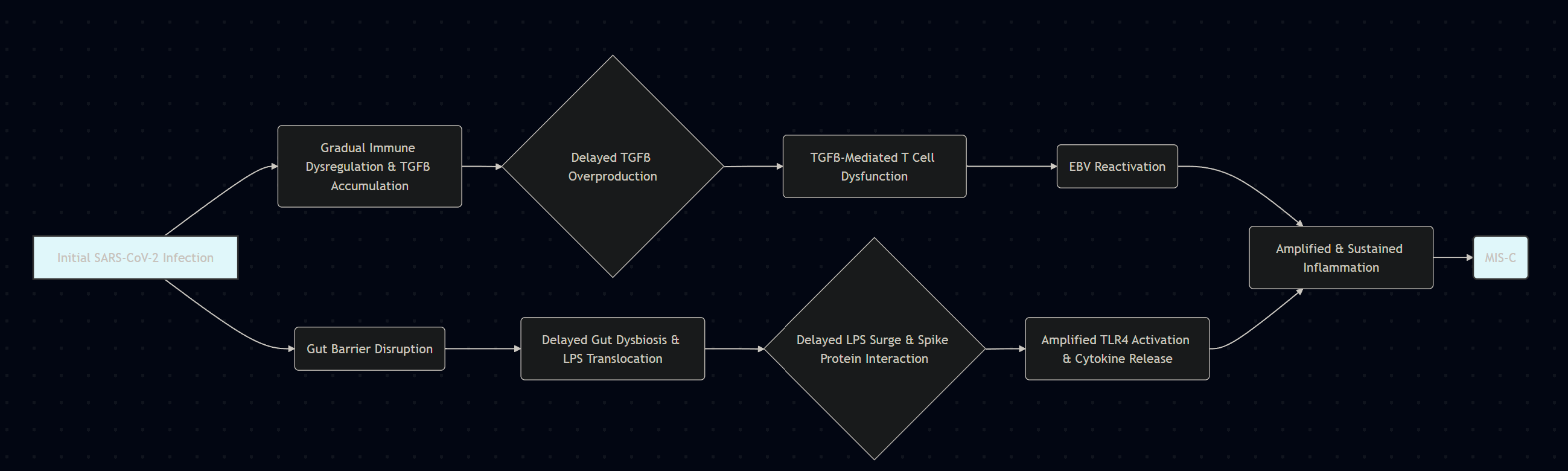
The initial inability of the child's body to properly degrade antigens from SARS-CoV-2, or its now well-known annoying persistence, may allow for not only one, but multiple pathways for a delay, hypersensitive reaction.
The road so far
Early in the pandemic, a literal handful of independent researchers from all walks of life were doing a lot of the heavy lifting, and thus I often looked where others didn’t, covering perspectives and pathways that had little attention. One of the pathways covered by a brilliant doctor named Theo was the TGFβ , thus why I never wrote much about it. He is gone from the internet, sadly, a massive loss, and so this timely paper started by path towards TGFβ.
This protein and its related pathway play a significant role in many of the more nuanced and paradoxical pathways I covered, it is central in immunosuppression, endotoxic tolerance, thrombosis, microvascular damage, it overlaps with Galectin-9 (which Omicron uses to “eat away” the gut protective layer), the so on.
Consider becoming a supporter, and if you do, or you are one, thank you very much !

Greetings Moriarty.
TGFβ search on PubMed returns a whopping 121,572 peer reviewed papers!
Of those 1,830 papers mention Endotoxin.
2,356 papers pop up with LPS
2,705 with Lipopolysaccharide
Injecting people with dead Bacteria predictably increases TGFβ with variation between individuals.
https://geoffpain.substack.com/p/endotoxin-jabbing-recommended-by
I am still dealing with things, so the next article may be delayed (this article wasn't delayed =D).
Integrins are the bane of my existence T_T.
Edit - Oh forget that, I may publish something tomorrow lol