
Do SARS-CoV-2 variants still bind to Endotoxin ?
Is it still relevant ?

Before we go through this highly impactful paper, I should refresh your memory, given how long ago we covered this topic in a detailed manner.

In my pursuit to understand what the virus and its highly toxic fragments could do, I sought outlier explanations as nothing else would fit or explain the untold amount of “oddities” in the data. What I thought at the time to be merely a conspiracy-minded “thought” turned out to be scientifically true.
SARS-CoV-2 Spike Protein binds to bacterial toxins and, in turn, can turn the cells into inflammatory furnaces.
The implications go far beyond hyperinflammatory responses, the infamous cytokine storm, which, in simpler terms, is when your body surges the production of inflammatory proteins and mediators, damaging organs systematically, overwhelming everything. Endotoxin is one of the biggest contributors to human disease, from neurodegeneration to cardiovascular diseases, and the general burden on multiple organs over time. Even at the cellular level, it can cause immune cells to respond more slowly to any subsequent threat (a process called endotoxin tolerance).
It is why patients who survive sepsis and septic shock are found to be susceptible to new infections years after the fact. A process called Immune Paralysis. It opens the doors for opportunistic pathogens. It directly shifts the cell metabolism towards the Kynurenine Pathway. It forms immunological scarring. The list goes on, and it is extensive.
Since then, some debate has occurred in the two communities. In the scientific community, many researchers argued that the virus mutated so much, especially its Spike Protein, the findings, even if valid, likely had no relevance. On the clinical side, an untold number of clinicians argued it is not clinically relevant for the same reason, and because medicine mostly researches severe damage, and such.
To me, not only was it clinically and scientifically relevant, but I argued it partially held a lot of importance to understand a large part of, well, everything.
Conservation of Bacterial Lipopolysaccharide Binding by SARS-CoV-2 Spike across Major Viral Variants
This paper is very technical, and at least to me, making protein interaction easy to understand is difficult. It is intuitively easy to learn, but difficult to explain, so I will do my best, because the topic is that important.
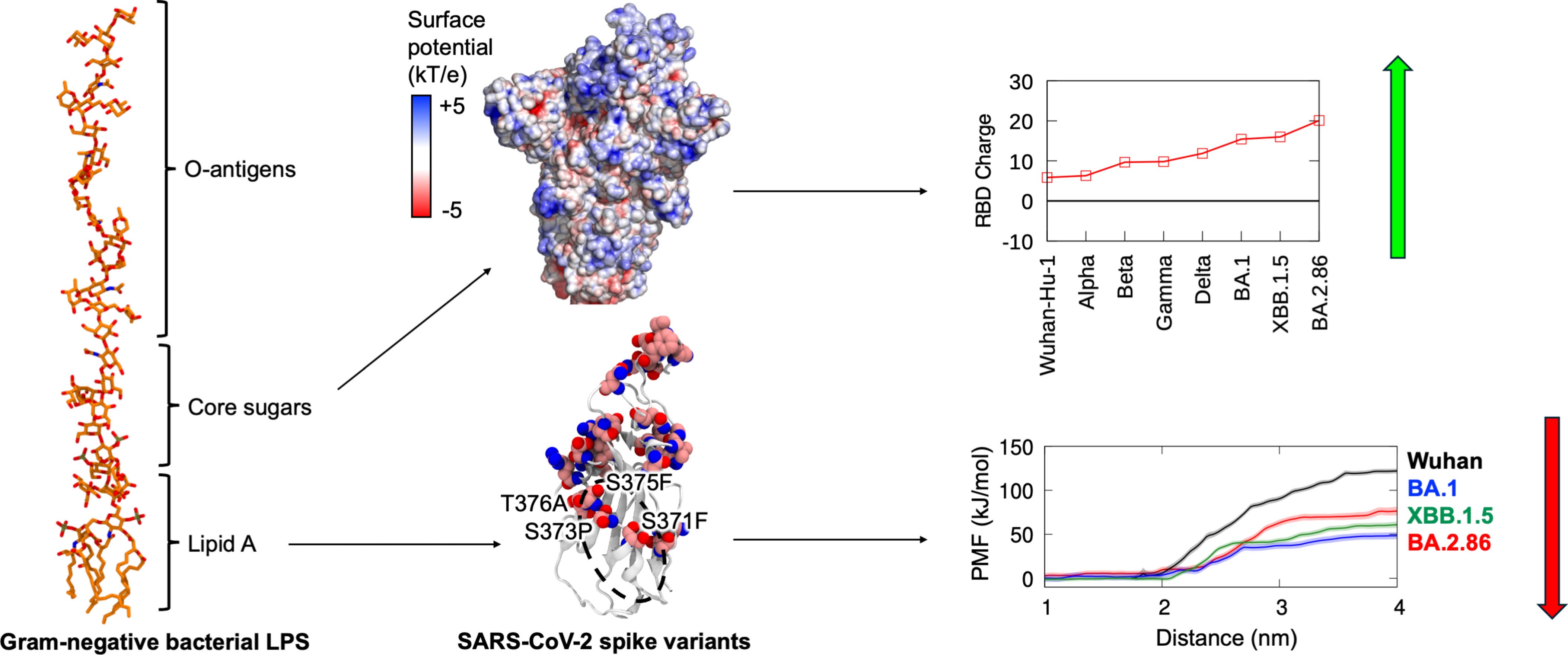
For contextual understanding, even singular mutations at precise places in the virus can completely change how the virus “behaves”. A mutation at one specific section of the virus can enable it to bind to one receptor better than others, alter which cells it can infect, and another mutation at another spot can enable it to evade a large part of your immune response. As cited by the authors, while all the non-Omicron variants, Alpha, Beta, Gamma, and Delta, gained roughly around 10 mutations, many Omicron variants present over 30 mutations.
Another example is the Omicron variant from mid-2023 (BA.2.86), possessing almost 50 mutations compared to the original Wuhan variant. With so many mutations, the rationale was that they abolished the Spike Protein's promiscuous binding to Endotoxin. Here, the authors decided to focus on the variants BA.1, XBB.1.5, and BA.2.86, which span a 4-year period of viral evolution, to test if the binding of Endotoxin was completely abolished, minimal, or changed.
The Spike Protein has many regions of significance. One of my main focuses for a long time has been the N-Terminal Domain, because of its paradoxical multi-functional aspect. The NTD has a specific region that mimics one of our most important proteins, Galectin-3, a protein that basically orchestrates highly complex inflammatory and immunological cascades. It resembles one of the infamous “HIV inserts” (but it arose naturally), and it is also an immunological supersite, our antibodies seek it with a vengeance. And crucially, it binds to endotoxin.
Because of its antigenic supersite nature, it will naturally bear many mutations, but most are located in regions distant from the LPS-binding pocket. On the other hand, the Receptor Binding Domain (RBD), also a highly sought-after antibody target, acquired mutations directly in its LPS-binding site. 3 mutations in BA.1 S371L, S373P, and S375F and 4 mutations in BA.2.86 and XBB.1.5 S371F, S373P, S375F, and T376A. Because these mutations are located at the heart of the highest-affinity LPS binding sites, the authors focused their investigation on the RBD.
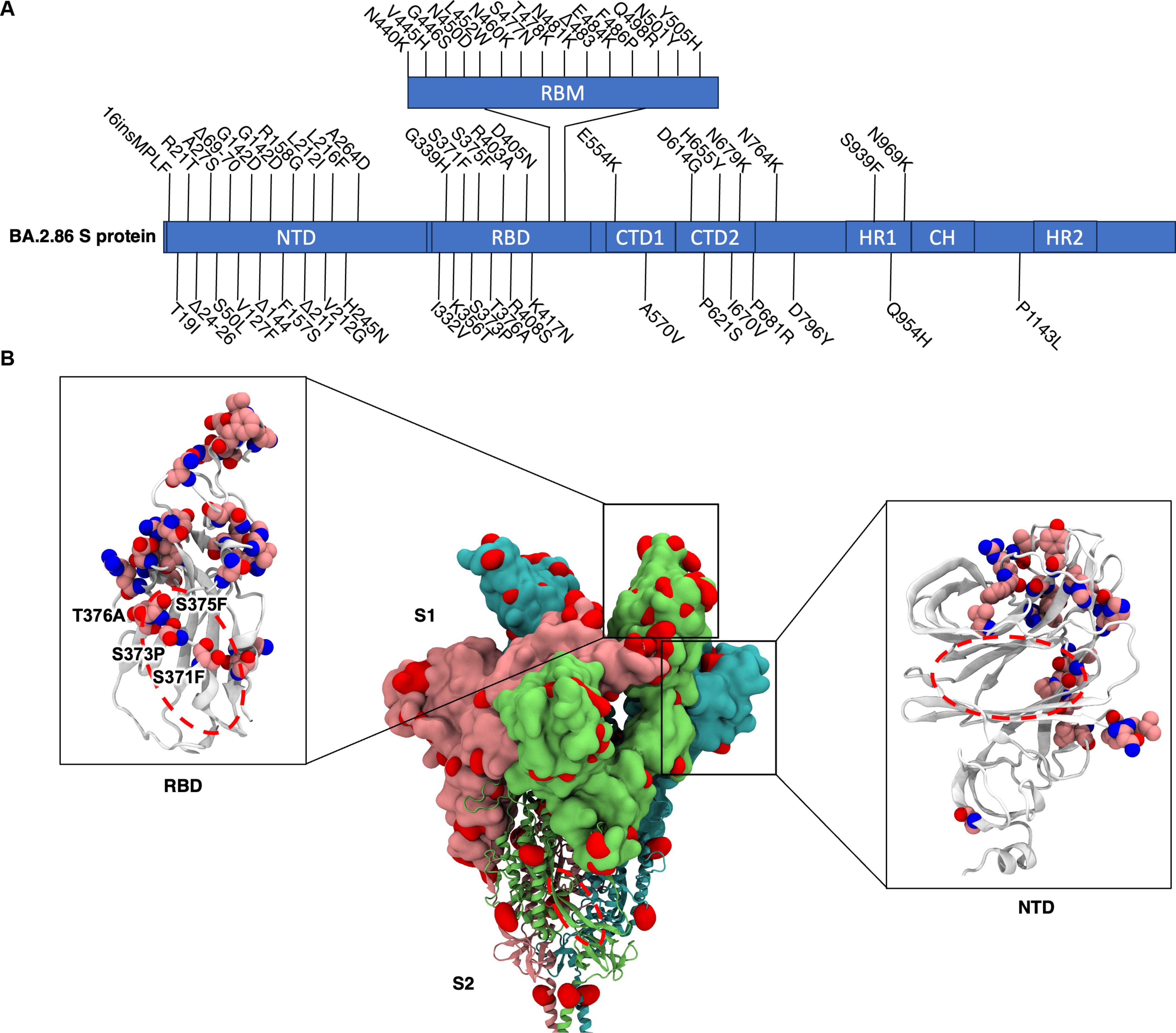
These positions are critical because, in the ancestral Wuhan-Hu-1 strain, the serine residues at 371, 373, and 375 form hydrogen bonds with the β-1,6′-linked glucosamine moieties of the lipid A (the most toxic part of LPS) headgroup. These bonds help stabilise the open conformation of the pocket and anchor the lipid. Mutating these serines to bulkier, nonpolar residues, such as leucine, proline, phenylalanine, or changing the adjacent threonine to alanine, would be expected to sever that hydrogen-bonding network and distort the helix that controls access to the pocket.
To test their hypothesis, they use a very precise test called Blue Native PAGE (A in the image below). This technique keeps proteins in their native state, maintaining their shape and allowing them to interact with other molecules as they would in a physiological context.
It measures how these interactions alter the protein’s migration through the gel. When they incubated the RBD from Wuhan, BA.1, XBB.1.5 and BA.2.86 with increasing concentrations of E. Coli’s LPS, all four variants showed a concentration-dependent shift in migration. Even when the RBD is intrinsically clumped into different states (oligomeric states), LPS still bound and altered their migration patterns. The dose-dependent shift in migration confirms that LPS binding occurs across all variants, regardless of their differing baseline “clumping”
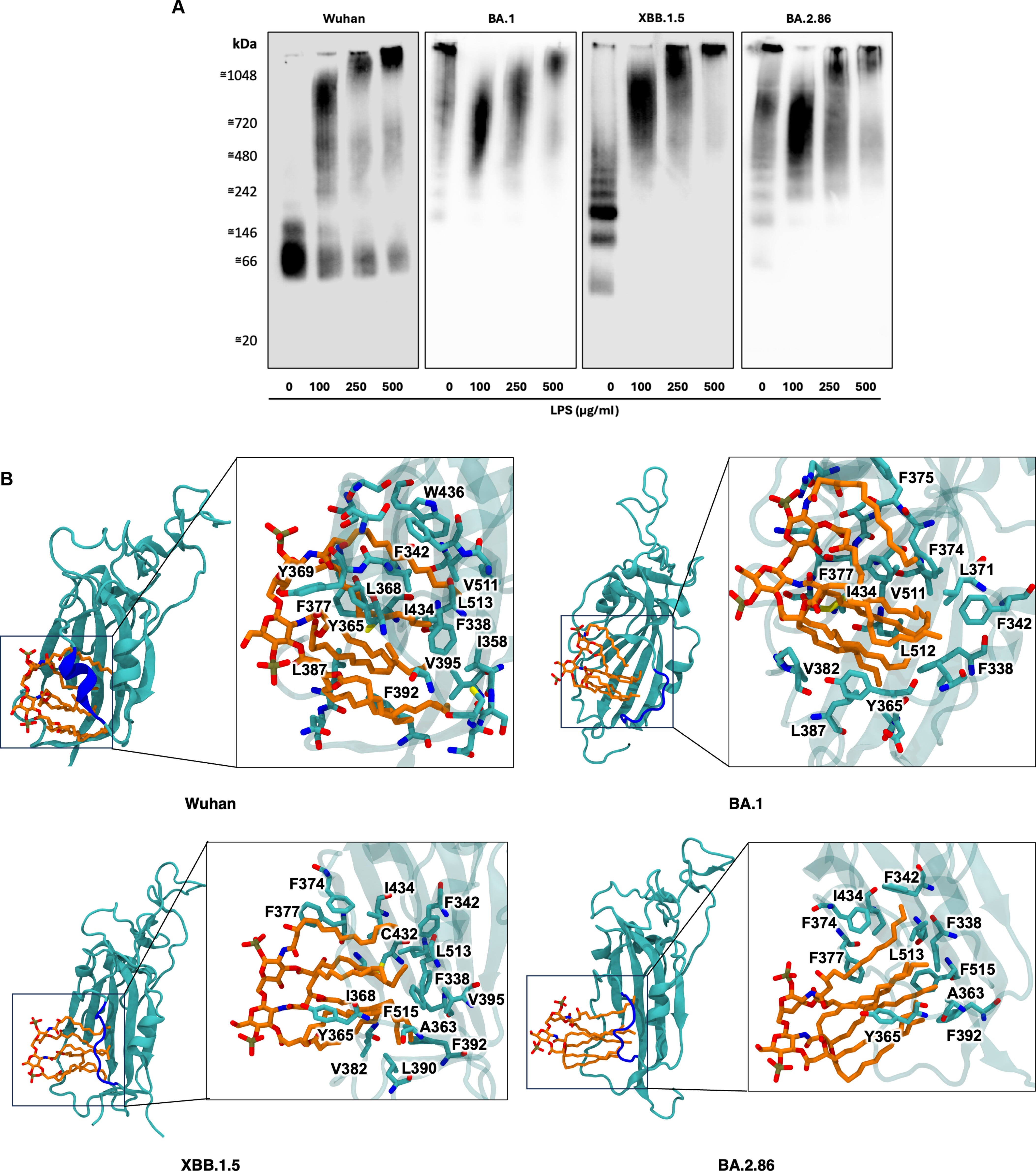
Computer simulations in bioinformatics and biophysics are extremely powerful tools, some of the most impactful papers usually have bioinformatics with well-designed laboratory tests, they are predictive by their nature. So, to understand the mechanism of the LPS to bind to mutated RBDs, that is what they did. For XBB.1.5 and BA.2.86 RBDs, they placed the Lipid A molecule approximately 2nm from the cryptic pocket and ran three independent 1-microsecond simulations per variant.
In every simulation, lipid A spontaneously bound to the cryptic (meaning hidden) hydrophobic pockets within the first 100 nanoseconds and remained stably bound throughout the remainder of the simulations. Despite all mutations in regions of interest towards Endotoxin binding (gating helix), the virus maintains that capability. These hydrophobic pockets are conserved across all variants.
Upon further inspection, they observed a structural distortion in the gating helix region (residues 364-371, blue in the image above). The gating helix, per the name, acts as a gate towards one of the hidden pockets of the RBD in the Omicron variants compared to the Wuhan variant. This is critically important as changes here imply a change in shape, resulting in the loss of the optimal shape to bind to Lipid A.
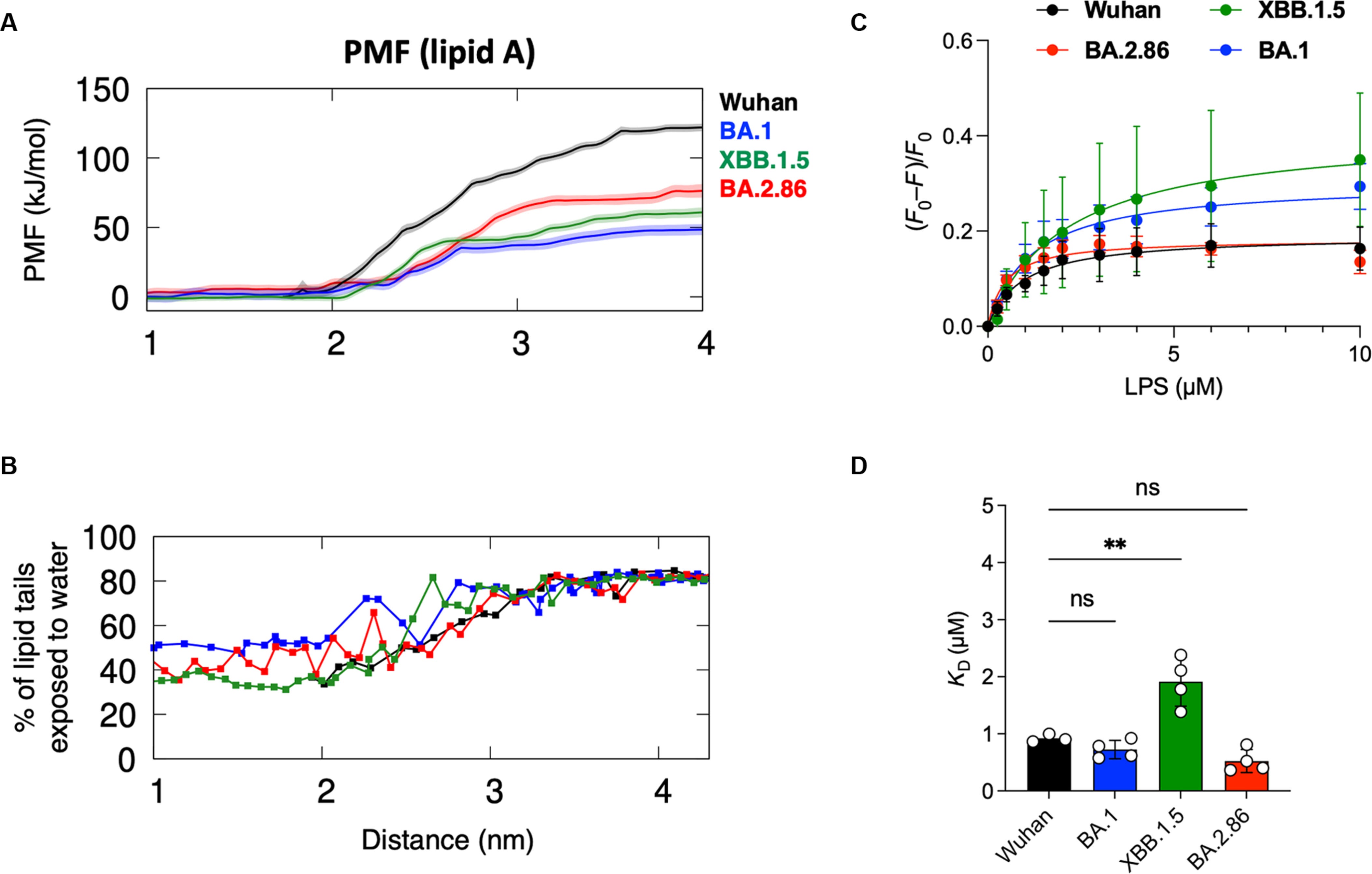
Due to this distortion, they hypothesized that lipid A binding in the XBB.1.5 and BA.2.86 variants would be weaker compared to Wuhan. They proceed to use steered molecular dynamics followed by umbrella sampling to calculate the potential of mean force (PMF). In simple terms, it is a test to quantify how strongly lipid A binds to the RBD, and this is a computationally expensive approach but highly precise.
As one would expect, the results demonstrated echoed the structural observations above. Wuhan has the strongest binding (125 kJ/mol), XBB.1.5 had 60 kJ/mol, BA.2.86 had 75 kJ/mol, and lastly BA.1 had the biggest reduction at 50 kJ/mol. This loss of Lipid A binding strength occurs likely because of the gating helix distortion, which cannot shield the hydrophobic pockets from water (something the Wuhan RBD did).
But if that is the case, why is severe Covid still associated with hyperinflammation, and why are bacterial infections still abundant ? The list of questions regarding the role of Endotoxin in Covid would diminish, not grow larger. To answer these questions and validate the computer modelling predictions, they performed fluorescence quenching experiments using full-length E. Coli LPS, not just its isolated lipid A. This technique measures how the tryptophan in the RBD loses fluorescence as LPS binds to it, allowing quantification of binding affinity.
For the XBB.1.5 variant, higher doses of LPS were required to achieve the same level of fluorescence quenching compared to Wuhan, consistent with the prediction, possessing a significantly weaker binding. Unexpectedly, BA.1 and BA.2.86 produced quenching comparable to Wuhan with similar LPS doses, basically no significantly different from the ancestral strain. Full-length LPS was binding just as tightly as before, even though the lipid A tail component was binding more weakly. How ?
E. Coli endotoxin is a complex molecule, consisting of 3 domains, the O-antigen made of highly variable, but repetitive polysaccharides, connected to the core oligosaccharide, a series of sugar molecules (core oligosaccharides, consisting of keto-deoxyoctulosonate sugars, heptoses, and hexones), followed by Lipid A. The core of E. coli contains a conserved inner core with two phosphorylated mannoheptoses, and this increases the overall charge of full-length LPS −10 e compared to −4 e for lipid A.
The long string of negatively charged sugars is likely to interact with the RBD surface outside the hidden pocket, providing a compensatory binding mechanism. They measure this by electrostatic potential. Full-length spike protein charge showed an overall increase in positive charge over time, but with distinctions. The Gamma variant and the XBB1.5 had lower charges than the preceding Beta and BA.1 variants. The changes in charge are not universal or uniform.
The NTD showed no clear trend, its charge fluctuated across variants, and the S2 subunit (which also binds to LPS) was similar. RBD charge, however, had a steady increase, from +6e in the Wuhan variant, to +10e on Alpha, to +15e on BA.1, +16e on XBB.1.5, to +20e in the BA.286. The authors identified up to six additional basic residues (arginine and lysine) in the RBD surface of the emerging variants compared to the Wuhan strain, these residues cluster in the regions accessible to the LPS sugar chain.
To observe this directly, they simulated full-length Wuhan RBD bound to rough LPS (lacking the O-antigen part), and smooth LPS (with O-antigen from E. Coli O111). While the lipid A tail remained stably buried in the pocked, the sugars were highly flexible, bending towards the RBD surface. Contact analysis revealed that the core sugars interact with positively charged residues near the pocket.
To enhance sampling of these highly mobile sugars, the authors ran additional simulations at 500K (with backbone restraints to preserve protein structure). Under these conditions, the inner core phosphorylated sugars consistently formed salt bridges with K378 and R408. These electrostatic interactions represent the most prominent binding force between the sugar chain and the RBD surface.
Mutations that distort the gating helix compromise lipid A binding, in turn, the accumulation of positive surface charge on the RBD enhances interaction with the negatively charged LPS sugar groups. It is likely a compensatory mechanism to conserve LPS binding through distinct mechanisms. Loss of hydrophobic pockets is supplemented by increasingly strong electrostatic interaction with sugar chains.
This matters. A lot.
SARS-CoV-2 Omicron BA.2.86 and JN.1 expand tropism in human proximal intestinal epithelium
Omicron SARS-CoV-2 has diversified into multiple sub-lineages, complicating assessment of their intrinsic phenotypes due to background population immunity. We compare replication and biological characteristics of variants from BA.1 to JN.1 using human bronchial and lung explants, airway organoids, colon cells, and proximal intestinal enteroids. XBB.1.5 and EG.5.1 achieve higher replication titres in respiratory tissues than BA.2.86 and JN.1, indicating enhanced respiratory fitness. EG.5.1 displays dual cell-entry pathways and greater replication in alveolar epithelial cells, supporting increased lung tropism and pathogenicity. In contrast, BA.2.86 and JN.1 rely on TMPRSS2-mediated entry in airways. Notably, BA.2.86 and JN.1 replicate more efficiently than EG.5.1 in proximal intestinal enteroids in an ACE2- and TMPRSS2-dependent manner, but not in colon cells. JN.1 exhibits elevated intestinal tropism with limited proinflammatory cytokine induction, suggesting potential for faecal transmission. Here we show XBB.1.5 and EG.5.1 greater transmissibility and severity potential whereas BA.2.86 and JN.1 exhibit enhanced intestinal adaptation.
I have argued for a long time that each variant has a preferred tropism. Similar to how mutations changed how parts of the Spike Protein can bind to Endotoxin, mutations will also change viral tropism, meaning the preferred “place” the virus seeks to infect. Infection of the gut has profound effects, even on mild infections, because the virus is very aggressive and replicates so fast that it will shift the microbiome, depleting many beneficial microbes. In turn, this allows bad ones to grow, but primarily, it thins the gut lining and allows Endotoxin to translocate.
The viral interaction with endotoxin causes a cascade of effects. It triggers both Spike Protein aggregation and amyloid formation. It can trigger the activation of Mast cells, it recruits and activates neutrophils, which, as we have seen, creates extremely toxic viral fragments. It can induce the formation of extracellular traps.
Ironically enough, the same cells that the toxic fragments (xenoAMPs) kill, are the ones necessary to control Endotoxin-driven inflammation. Neutrophils themselves are responsible for the mitigation of inflammation derived from endotoxin. So the very cell responsible for clearing clumps of Spike Protein+LPS generates fragments, which themselves allow LPS to bypass defence mechanisms and act inside the cell. The graph at the end of my last article about Coronaviruses activating mast cells can give you a great overview of the multiple mechanisms.
We are now having glimpses of how and why viral fragments are lasting significantly longer than most other viruses, as the body is simply taxed by opposing and also paradoxical stimuli. My focus is not hyperinflammation, or acute damage, but long-term consequences, the well-known death by a thousand cuts.
Addressing gut health and gut lining is very individualised, but addressing endotoxin exposure is much simpler.
You can use natural fibres, which will “soak in” endotoxin from the gut, with oat and wheat bran being highly effective (you can find me suggesting wheat bran to clean the gut from the virus and its viral particles some 2-3 years ago). If that is not an option or simply something you don’t want to do, activated charcoal is extremely effective and usually cheap.
Before ending this article, I would like to add something (which I usually would add in the comment section), a quote directly from the authors.
The reduction of complexity to an isolated RBD in this study was necessary here to allow for the extensive free energy calculations across many variants and large-scale simulations with full-length LPS, which would otherwise be prohibitively expensive if the S protein trimer or even the S1 subunit was used.
It is something that usually flees my mind. I often forget how absurdly expensive many of the observations I make, and tests I would like to do (hinted at throughout articles) actually are. The computational costs alone would be prohibitively expensive, let alone some of the laboratory costs, which would involve designing new tests altogether. So I found it refreshing reading this from a group of researchers, a reminder to myself.
Consider becoming a paid subscriber, and I appreciate the patience of those who continue to support my work.
You can also do a one-time thing by buying me a coffee.

I hope all of you have been well. I have been... something. Regardless, I may do a rapid-fire sequence of articles.
Shorter in nature to cover a lot more ground, given how extensively I have broken down multiple mechanisms, there is a lot of new information that you should know.
Secondly, I do not remember if I ever even once republished an article, perhaps once, but I will likely do it in the next few weeks with two articles, as I find myself in the position to continue a specific line of research (not related to SARS-CoV-2... but with Language).
See you soon, hopefully.
COVID-19 varients:
Alpha
Beta
Delta
Gamma
Epison
Theta
Count me confused, is 'Moriatry' a brain wave as well? Must be.