
Persistent CD4⁺ T Cell Dysfunction After Mild COVID-19
Neo polymorphic toxin indeed

This will be a standalone piece, but as with other articles lately, for a thorough understanding of the subject, and especially to understand my overall hypothesis, additional reading or having notes is advised. Validation of fringe (as in the cutting-edge of science) often takes time, and this one is central to one of the cores of my hypothesis.
Close to my birthday in December of 2024, I finally wrote a presentation on one of my core hypotheses. SARS-CoV-2 and especially its Spike Protein act as a neo (novel, new form) of a polymorphic toxin, this NPT subverts endogenous toxins from different pathogen species to its own use, not merely by mimicking them, which also does.
I have argued consistently that SARS-CoV-2 not merely mimics one of the world’s most powerful toxins that acts as a superantigen, it actively subverts and creates entire reactionary cascades as it acts as one by itself.
The functional immune deficiency I referred to recently, which occurs not only after a severe infection, but through longer timeframers would be a byproduct of this novel behavior. Per usual, I will first break down the paper, then proceed to connect to older articles/work and what the findings implicate.
Persistent CD4+ T cell functional deficits during recovery from prolonged symptomatic SARS-CoV-2 infection
The authors focus in this paper, per the title, is the persistent CD4+ T Cell dysfunction experienced during recovery from mild to moderate infection, with no hospitalization. This distinction in the data is very important because the more severe an infection, especially from earlier strains, the deeper the scars in the body’s biology. Mild to moderate infection data can be extrapolated to a wider population, with a bit of care.
The analysis of CD4+ T-Cells is important because CD4+ T-Cells are critical for orchestrating systemic immunological responses, both from other T-Helper cells (1, 2, 17, fh) but also CD8+ (killer cells), macrophages (eaters of pathogens, and other things), and B-Cells (antibody responses). Dysfunction in any form in these cells and their receptors makes one susceptible to secondary infection with ease, and is also central to controlling chronic infections and reactivation, such as herpes.
The samples were collected pre-vaccination, which is a point the authors bring up. The samples came from 17 people of both genders between 26 and 71 years old. “Seven participants were studied at one time point, nine were studied at two time points, and one was studied at four time points. The time between blood draws ranged from 63 to 178 days. Three participants (01, 03, and 04) received their first COVID-19 vaccination before the second visit.”
Participant 04, who showed the most delayed recovery, was among those vaccinated before the second draw, remaining CD4 low at 8 months and only beginning recovery at 11 months. Control samples were all pre-2019 to ensure no SARS-CoV-2 exposure.
The authors purposefully used Staphylococcal Enterotoxin B (SEB) as a means to test activation and measure numerous markers and proteins in the samples because it is a superantigen that bypasses both antigen-specificity (thus it test T-Cell Receptor signaling itself, not just SARS-CoV-2 responses), it engages an absurd amount of T-Cells, and also the Spike Protein has not only 1, but 2 SEB-like sequences, with the second one being even more potent than the first.
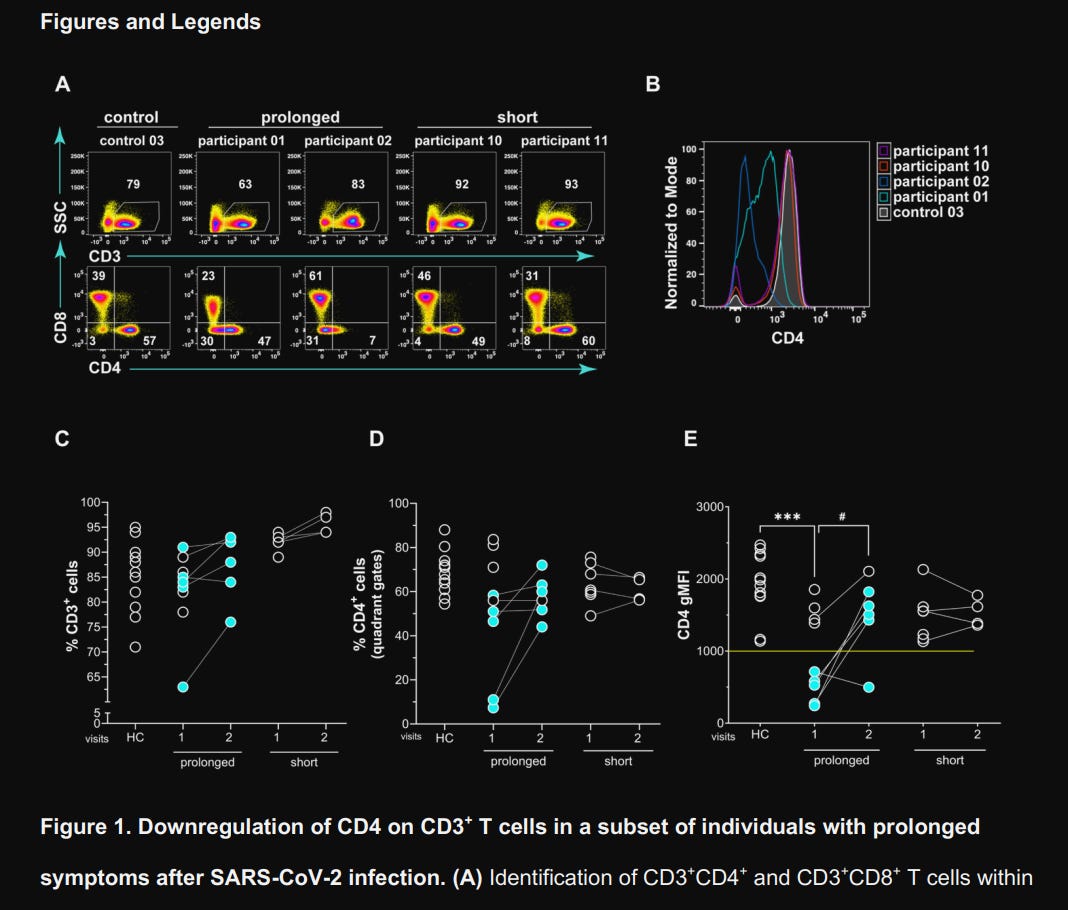
They use a more precise form to measure not only the percentage but the density of CD4 itself (per cell surface), called “Geometric Mean Fluorescence Intensity” (gMFI), and found significant downregulation of CD4 on CD3+CD8– T cells (the CD4+ T cell population) in 5 of 9 patients experiencing prolonged symptoms. Among these 5, 4 showed increased CD4 expression over 3 to 12 months, but patient 4 remained CD4low even at 8 months post-infection, only starting recovery at 11 months. The short-term symptom duration group had normal levels throughout the test.
These effects were not related to lymphopenia (death of these immune cells), given that they were present in normal amounts. It is because they were low in said receptor (CD4). To assess if this altered their functional capacity, independent of SARS-CoV-2 specificity, they stimulated the blood cell samples of the participants and their healthy control counterparts with SEB.
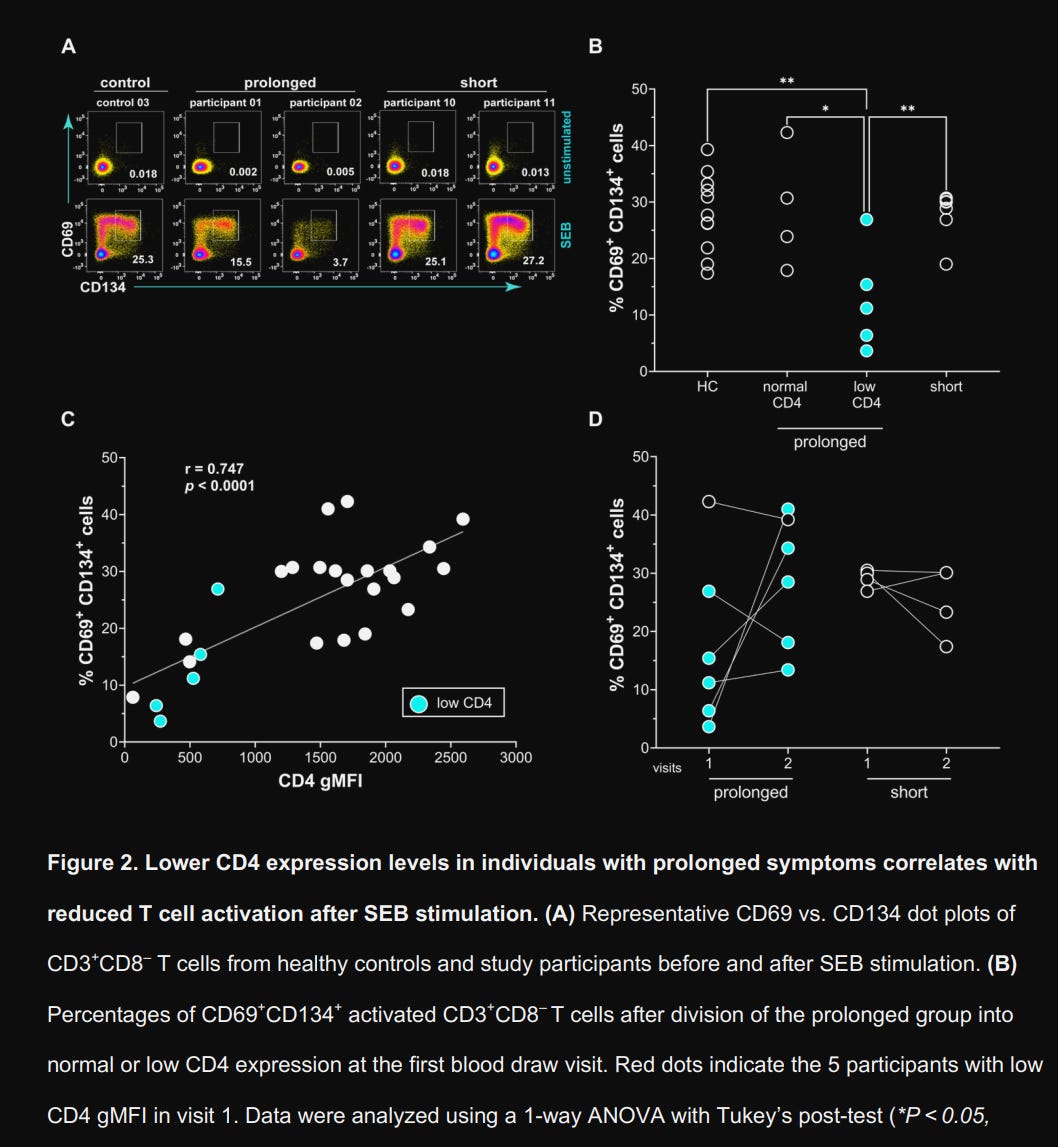
The CD4 low group exhibited profound hyporesponsiveness to SEB, with only 12.7% of cells in that group showing activation (CD69+CD134+) compared to 28.1% in healthy controls, with a strong positive correlation (r = 0.74) with the gMFI measurements above. After CD4 levels normalized in longitudinal samples, responsiveness recovered, jumping from 9.17% to 29.3%. Does this translate into meaningful systemic changes ?
Yes. They analyzed 52 different cytokines and found an inverse correlation between CD4 expression and critical inflammatory markers. IL-1RA (antagonist, lowers IL-1), was elevated, suggesting active IL-1β pathway inhibition, indicating ongoing Type I inflammation despite apparent recovery.
IL-7 was paradoxically elevated in CD4 low patients, and as we know, this interleukin acts as a survival and proliferation signal to T-Cells, a compensatory homeostatic response attempting to maintain T cell numbers despite their dysfunction.
Remarkably, they also found increased Vascular Endothelial Growth Factor (VEGF), and this is a clear sign of ongoing vascular inflammation and endothelial distress. Anti-SARS-CoV-2 S1 IgG titers showed no correlation with CD4 expression, indicating the T cell defect is not a consequence of inadequate humoral response or persistent high viral antigen load alone.
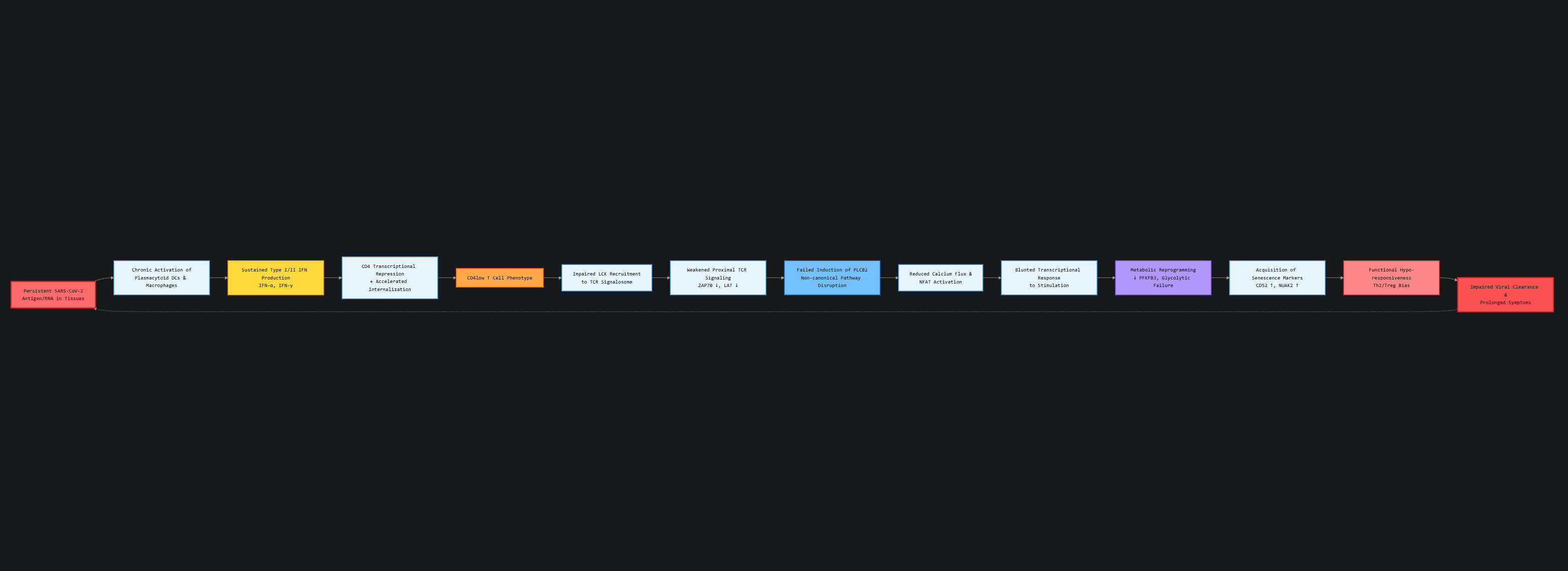
As with some of my articles in recent months, I have stated that analyzing the different genes—which ones go up, go down, or are differently expressed can give you deeper insight and a path towards answers and effective treatments. The CD4 low group showed a 56–57% reduction in differentially expressed genes (DEGs) following SEB stimulation compared to healthy controls and recovered samples, demonstrating a global activation defect, meaning the T-Cells are not just responding (skewed) in one way, they harbor systemic blunted transcriptional responses upon their receptor being engaged.
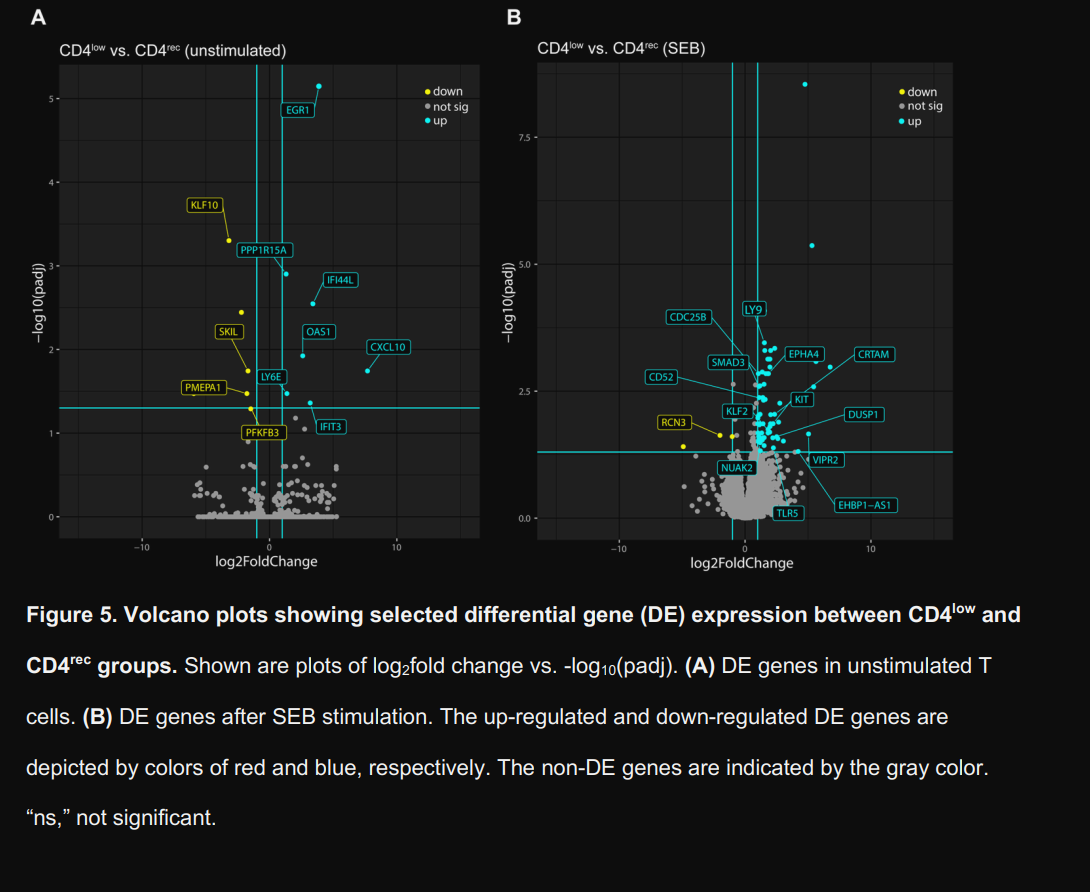
To have an even more detailed picture, they analyzed upstream regulator genes and pathways of interest and compared the changes between CD4 low and CD recovery, on both unstimulated and SEB-stimulated cells. Unstimulated CD4 low cells show increased expression of Interferon-stimulated genes, such as IFI44L, OAS1, IFIT3, and CXCL10, and among other roles, they are primary drivers of IFN-γ responses.
While the authors focus here is on a broader chronic stimulation leading to lower expression of CD4, I would like to add that IFN-γ itself is actively driving the gene expression pattern here. IFN-γ itself can induce CD4 internalization, causing remarkably lower expression in T-Cells.
They also note an upregulation of Early Growth Response Factor 1 (EGR1), which is usually observed in Th2 (allergic) responses. Suppressor of Cytokine Signaling 1 (SOCS1) was predicted to be inhibited. Per its name, SOCS1 acts as a negative feedback to dampen excessive or chronic inflammation, and being inhibited allows inflammation to persist and drive further changes, creating an inflammatory feedback loop.
These genes are chronically active and enriched, suggesting that the cells have been inundated with interferons, and this is evidence that these effects occur via viral fragments, their persistence, and also bystander activation via inflammation, lasting months. It aligns with the evidence that the body takes months to clear the viral remnants of one single infection.
Upon using Ingenuity Pathway Analysis (IPA), a software that analyzes and integrates multiple data sources (such as measuring RNA, genes, proteins, etc) to do complex analysis and predict pathways, they found what was described as part of the “Hypercytokinemia/hyperchemokinemia in the pathogenesis of influenza”.
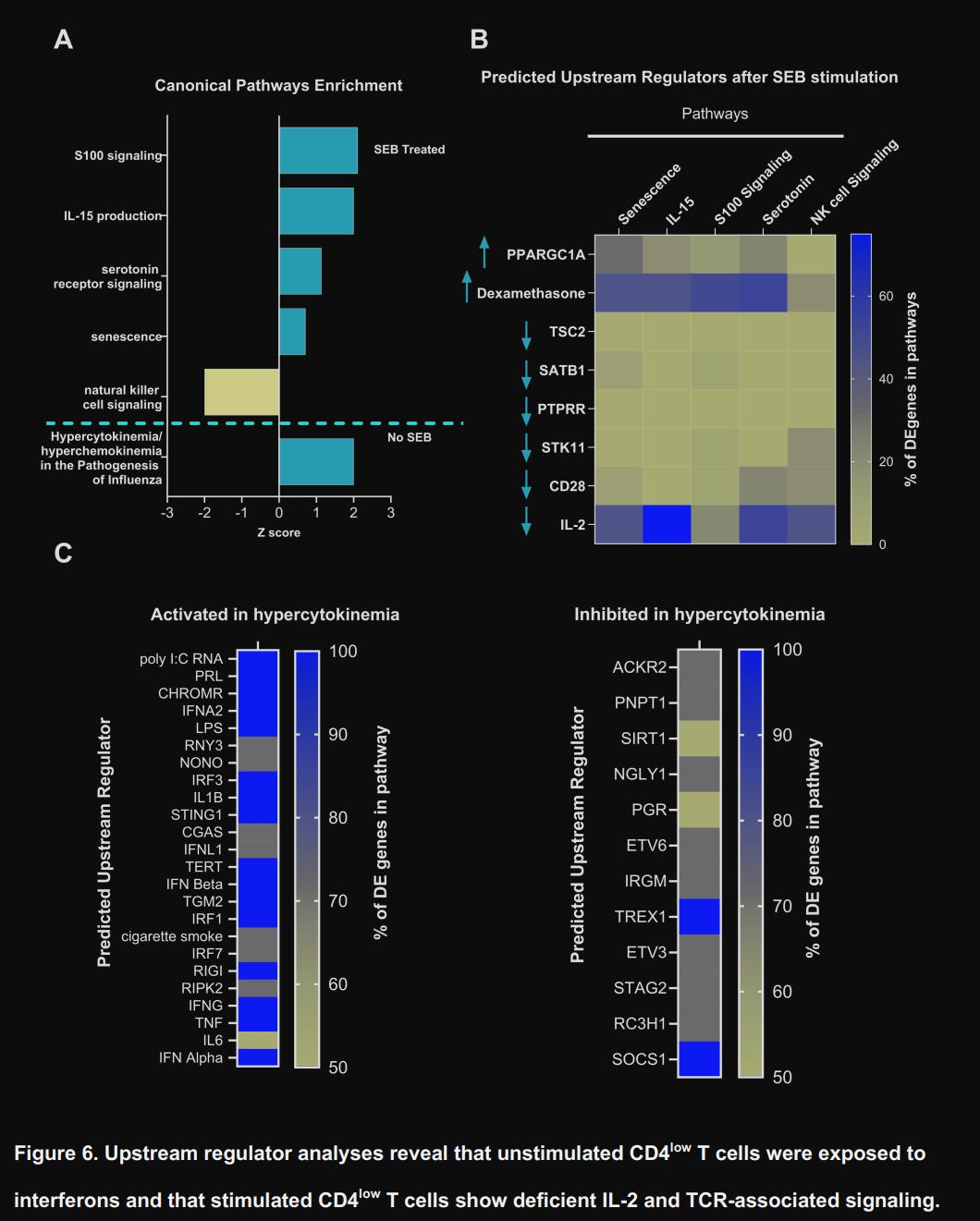
However, upon stimulating CD4low T-Cells with SEB, the pathways changed dramatically. Genes related to the pathways “S100 signaling” (inflammation/calcium signaling), “IL-15 production” (homeostatic survival), “Serotonin receptor signaling” (neuro-immune modulation), and, critically, senescence were all increased. On the other side, they found striking downregulation of Natural Killer Cell signaling, IL-2, and CD28, which are essential for full T-Cell activation, proliferation, and effector function.
One of the most remarkable mechanistic findings in the paper was the defect of PLCB2 expression (which goes back to normal in CD4 recovered), critical to both signaling and amplifying immune response via the alternative superantigen pathway, but also controlling cellular metabolism.
Also found downregulated was PFKFB3, a master regulator of glycolysis, the metabolic pathway T cells rely upon for rapid proliferation and effector function. Its downregulation impairs proper energy generation, essentially starving the cells of ATP when they need it most.
Together with upregulated markers such as CD52 and NUAK2, both associated with suppression of T cell activation and induction of senescence, with no elevation of common exhaustion markers (PD-1, TIM-3, LAG-3, or TIGIT), these cells are in a senescence (zombie) like state. Not exhausted, not dead, but metabolically paralyzed, inert.
What does this all mean on its own?
After initial infection, T-Cells with low expression of CD4 are found to be in a paradoxical hyporesponsive state, with profound metabolic reprogramming (they can’t generate or use energy efficiently), and they don’t respond to incoming dangers despite being in a chronically activated inflammatory milieu.
There is also an indication that the body is actively pushing these cells toward becoming regulatory T-Cells (via SMAD3 upregulation and SKIL downregulation), perhaps as a desperate attempt to throttle this inflammatory loop, effectively sacrificing effective immune defense for survival.
Worth noting that while the CD4 recover their proper function, and normal expression of many markers, they are not the same as healthy individuals, so even a mild symptomatic infection will leave scars in your immune system.
Paradoxically Acquired Immune Dysfunction
Unbeknownst to the authors, they executed cutting-edge research and gave me a lot of data and information to corroborate my earlier assertions and hypothesis proposal. My proposed framework, an attempt to integrate the rather unique molecular architecture of SARS-CoV-2 with its paradoxical sequelae, now has more ground to stand.
The CD4 low T-Cells that become hypo-responsive towards SEB stimulation, with a specific failure to upregulate PLCB2, is evidence that chronic exposure to viral fragments (after all, superantigens can remain in the body for longer than estimated previously), which causes active desensitization of immune responses through receptor downregulation and signaling inhibition.
In the IPA integrative analyses, they found an extremely particular signature, it is as if these cells were under dexamethasone treatment, which repression of PLCB2 transcription would act as a protective mechanism against superantigenic stimulation. T-Cells stop responding as a means to stop overresponding to most stimuli.
The IFN signature in CD4low T cells likely activates tryptophan catabolism via IDO. This depletes tryptophan, a known inhibitor of mTOR activity. Since PFKFB3 is mTOR-dependent, this could explain the metabolic defect (a hypothesis requiring direct testing of kynurenine levels and mTOR-PFKFB3 signaling in CD4 low T cells).
Per my Neopolymorphic Toxin proposal, many of the aspects of the sequelae present at all levels are similar to what is seen in surviving patients of Toxic Shock Syndrome (TSS), and the slow recovery of CD4 expression mirrors the slow recovery seen in TSS after survival.
This is evidenced by the cytokine signature. IL-1RA elevates to counteract IL-1β in toxic shock, IL-6 is known to drive its acute phrase, and VEGF induces vascular permeability and capillary damage, both characteristic of septic and toxic shock. Chronically, it induces endothelial dysfunction and tissue hypoxia, perpetuating the immunosuppressive microenvironment.
This and other recent article’s observations of elevated IL-7 are particularly significant in the context of NPT. IL-7 rises during lymphopenic stress, attempting to drive homeostatic proliferation. In TSS and severe sepsis, the initial lymphopenia (from T cell activation-induced cell death and trafficking to tissues) triggers compensatory IL-7 production.
The CD4low cells are attempting to survive in an environment of chronic endotoxemia amplified by superantigenic stimulation.
Years ago, I proposed that a lot of the sequelae observed after a SARS-CoV-2 infection were remarkably similar to both the immune paralysis seen after Toxic Shock and, in a similar vein, to Endotoxic Tolerance. In PAID, a paradoxical state of immune paralysis and immune dysfunction, there are opposite signs, but here are signs that PAID has significant ground to stand on. Tell me if any of these are true.
Post-Covid patients show increased rates of Fungal infection, such as Aspergillus, Mucormycosis
Herpes virus reactivation (EBV, CMV, VZV), due to loss of immune surveillance (CD4 low), walking sepsis without any signs (endotoxic tolerant states allow infection to progress without fever or inflammatory signs).
Kynurenine Pathway and its metabolites are present and abundant for extended periods
Vaccinated individuals would possess poor quality and low-affinity antibodies, even after infection
Impaired Memory B Cell formation
Poor CD8+ T Cell priming, all explaining breakthrough infections and poor protection.
Neurodegeneration
Chronic IFN-γ signaling induces sustained IDO1 expression, leading to local tryptophan depletion and kynurenine pathway activation. This engages amino-acid stress responses (GCN2) and suppresses mTORC1 activity, promoting translational arrest and metabolic reprogramming away from glycolysis. Glycolytic regulators such as PFKFB3 are downregulated, biasing cells toward quiescence, tolerance, under persistent stress, thus in a senescent state.
The list could go on, but in case you are asking yourself if any of these are true. They all are, and I wrote an article, or even multiple articles, on each. This is a universal mechanism, and per the paper data itself, the reason there is so much both overlapping and distinction in sequelae is individual genetics, epigenetics, nutritional status, and underlying conditions. Genetics and sequentially epigenetics will play a substantially larger role here.
While there is both direct and tangential evidence towards some of my “outlandish” ideas and hypotheses, this is the first time that we are presented with grounded evidence towards one of my core hypotheses, allowing for further research, better comprehension of what is going on, and thus enabling effective ways to deal with the kaleidoscope of sequelae that follows all degrees of infection.
I can now work on PAID and NTP as a central driver, so I will get back into this topic with time. Work and research like this take a lot of time and effort.
Thank you for your support. If you want to help my research, you can buy me a coffee as a one-time thing or become a paid subscriber. Both are very helpful.

Just out of curiosity was the status of vitamin D levels known? There are studies that show that vitamin d deficiencies impairs immune regulation so it would seem to follow that could be a contributing factor to T cell disfunction. Is it possible many of these people are also deficient in vitamin d compounding the effects of the virus? Has this been looked at? The vast majority of people are vitamin d deficient and also fail to have proper sun exposure.
Thank you JP
Zombie cells. Yes, we have a theory the LNPs are part of that story as well.