
Flu mRNA vaccine failure and a new road to autoimmunity
And superior natural immuntiy

I decided not to merely write a “dunking” substack, but elongate it and bring up serious issues with how world governments and pharmaceutical companies plan to tackle arising issues, but first things first, and most readers will be aware of this one by now.
Past SARS-CoV-2 infection protection against re-infection: a systematic review and meta-analysis
In this systematic review and meta-analysis, we identified, reviewed, and extracted from the scientific literature retrospective and prospective cohort studies and test-negative case-control studies published from inception up to Sept 31, 2022, that estimated the reduction in risk of COVID-19 among individuals with a past SARS-CoV-2 infection in comparison to those without a previous infection. We meta-analysed the effectiveness of past infection by outcome (infection, symptomatic disease, and severe disease), variant, and time since infection. We ran a Bayesian meta-regression to estimate the pooled estimates of protection. Risk-of-bias assessment was evaluated using the National Institutes of Health quality-assessment tools. The systematic review was PRISMA compliant and was registered with PROSPERO (number CRD42022303850).
Findings
We identified a total of 65 studies from 19 different countries. Our meta-analyses showed that protection from past infection and any symptomatic disease was high for ancestral, alpha, beta, and delta variants, but was substantially lower for the omicron BA.1 variant. Pooled effectiveness against re-infection by the omicron BA.1 variant was 45·3% (95% uncertainty interval [UI] 17·3–76·1) and 44·0% (26·5–65·0) against omicron BA.1 symptomatic disease. Mean pooled effectiveness was greater than 78% against severe disease (hospitalisation and death) for all variants, including omicron BA.1. Protection from re-infection from ancestral, alpha, and delta variants declined over time but remained at 78·6% (49·8–93·6) at 40 weeks. Protection against re-infection by the omicron BA.1 variant declined more rapidly and was estimated at 36·1% (24·4–51·3) at 40 weeks. On the other hand, protection against severe disease remained high for all variants, with 90·2% (69·7–97·5) for ancestral, alpha, and delta variants, and 88·9% (84·7–90·9) for omicron BA.1 at 40 weeks.
Interpretation
Protection from past infection against re-infection from pre-omicron variants was very high and remained high even after 40 weeks. Protection was substantially lower for the omicron BA.1 variant and declined more rapidly over time than protection against previous variants. Protection from severe disease was high for all variants. The immunity conferred by past infection should be weighed alongside protection from vaccination when assessing future disease burden from COVID-19, providing guidance on when individuals should be vaccinated, and designing policies that mandate vaccination for workers or restrict access, on the basis of immune status, to settings where the risk of transmission is high, such as travel and high-occupancy indoor settings.
As straightforward as a scientific paper can be, pretty simple. Doing meta-analysis (analyzing a bunch of specific data, in this case, other scientific papers) spanning a sizable portion of the pandemic, they found that protection from infection without vaccination is as good or superior to the protection from vaccination, and in fact, it is actually longer.
Since the first roll-out of the mRNA vaccines, and for most of this substack existence I have covered the evidence of the same, at many different levels. This presents us with a problem, a problem that has been growing in “evidence” and being named the last few months quite a lot. The problem of immune imprinting is something not only I and many others covered, but warned. Add the following to the growing long list of evidence of this effect. For brevity’s sake, I won’t list every single substack I wrote about this, but this would be the most recent one.

Antibody Response to Omicron BA.4–BA.5 Bivalent Booster
Boosting with new bivalent mRNA vaccines targeting both the BA.4–BA.5 variant and the D614G strain did not elicit a discernibly superior virus-neutralizing peak antibody response as compared with boosting with the original monovalent vaccines. Limitations of our study include the small sample size and follow-up period of our groups. We also note that the between-group comparisons were not controlled for factors such as age, vaccine type, and health status, which may have had an effect on antibody responses. These findings may be indicative of immunologic imprinting,5 although follow-up studies are needed to determine whether antibody responses will deviate over time, including after the administration of a second bivalent booster.
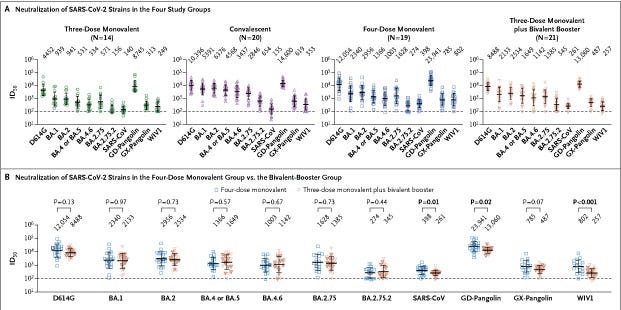
There is no discernible benefit to boosting with using a bilavent booster in comparison to monovalent, there is nothing groundbreaking here, just further evidence of a specific effect most is familiar with at this time. While it does sound like “beating a dead horse”, there is a specific reason I decided to take longer to publish this one was two-fold. First, the secondary mRNA failure mentioned in the title.
UPDATE 1-Moderna flu vaccine delivers mixed results in trial, shares fall
Moderna Inc on Thursday said its closely-watched experimental messenger RNA-based influenza vaccine generated a strong immune response against A strains of the flu but failed to show it was at least as effective as an approved vaccine versus less prevalent influenza B.
Moderna, whose only marketed product is its COVID-19 shot, has high hopes for its flu vaccine and aims to grab large portions of the respiratory syncytial virus (RSV) and seasonal flu markets with new mRNA vaccines.
The company said its vaccine, called mRNA-1010, generated a stronger immune response for the A/H3N2 and A/H1N1 strains than the marketed vaccine it was tested against in a trial of 6,102 adults aged 18 and over across Argentina, Australia, Colombia, Panama and the Philippines during flu season there.
It failed to meet its goal of non-inferiority compared to the conventional vaccine for the B/Victoria and B/Yamagata-lineage strains, the drugmaker said.
Moderna's shares fell more than 6% in after-hours trading following publication of the results.
The U.S. company said it has already updated mRNA-1010 in a way it believes will improve immune responses against Influenza B and will test those changes.
"We have always said our goal is to produce a flu vaccine, and then to iterate it, and to fine tune it over time to really make it exceptional," Chief Medical Officer Paul Burton said in an interview.
Dr. David Boulware, an infectious disease specialist at the University of Minnesota Medical School, said he was not overly concerned about the immune response versus Influenza B.
Boulware said the immune response against the A strains demonstrated that the vaccine probably worked and Moderna's tweaks to the vaccine are likely to improve the response against the B strains.
Seventy percent of those who received Moderna's shot reported mostly mild adverse reactions compared to 48% for the conventional flu vaccine. Pain and swelling at the injection site as well as headaches and fatigue were among the most commonly reported side effects.
The company also has an ongoing late-stage efficacy study on the mRNA-1010 flu vaccine, which could have data within weeks.
If that trial reads out soon, Burton said he hopes to have the data prepared and sent to regulators in the first half of this year, which could allow them to review it as soon as late 2023 or early 2024.
The flu, an infection of the nose, throat and lungs, kills 290,000 to 650,000 people worldwide annually. (Reporting by Patrick Wingrove and Michael Erman in New York; Editing by Bill Berkrot)
Moderna’s attempt at an Influenza mRNA vaccine failed to meet the expectations, it failed to meet the necessary levels for approval, especially against B strains. It has been a criticism of mine since the inception and massive hype on the mRNA, that outside a few pathogens and none being respiratory viruses, the mRNA wouldn’t work, especially against influenza viruses. If the mRNA against flu is anything but perfect it will turbocharge the influenza mutation rate, which is already something else.
Not exactly the point of bringing this up, but the following. For months I have warned here and there about cryptides, hidden peptides sequences that form unknown functional proteins, and effects on the body in regards to SARS-CoV-2. Here is a short substack in which I directly cite a paper about hidden peptides.
I highly recommend anyone interested in this subject to read the entire paper, even if it takes a few reads throughout the entire thing since the subject is really hard, but it will be incredibly beneficial as time goes on in my opinion.
The Hidden Enemy Within: Non-canonical Peptides in Virus-Induced Autoimmunity
One of the salient features of autoimmunity lies in the recognition of MHC class I associated self-peptides by T-cells that have escaped self-tolerance during their development and selection in the thymus. Self-peptides, not generated and presented at substantial levels for T-cells to undergo anergy and deletion, can cause T-cells to escape selection. Such cryptic peptides might be generated and presented under certain stimuli to activate self-specific T-cells, forming the fundamental basis of autoimmunity (Sercarz et al., 1993; Lanzavecchia, 1995). Over the years, many extrinsic factors including viral infections have served to explain autoimmunity through the generation of self-peptide mimics, bystander activation of T-cells and epitope spreading (Smatti et al., 2019). However, the exact nature and generation of such peptides remains loosely defined, hampering the development of broad range therapeutics and our understanding of these disorders.
Oligopeptides arising from the degradation of self and non-self-proteins enter the antigen presentation pathway, activating humoral and cell-mediated immune responses against infected and cancerous cells. While peptides derived from exogenous proteins, acquired externally through the endo-lysosomal pathway are loaded on MHC-II molecules, peptides derived from endogenously synthesized proteins are presented on MHC-I (Vyas et al., 2008). Such peptides form the basis of CD8+ T-cell immune responses that play an important role in various infections, tumors, and autoimmune disorders. However, efficient presentation of endogenous foreign peptides requires them to compete with both high and low-affinity self-peptides originating from the cellular proteome. This competition would be puzzling in viral infections, where slow synthesis and degradation of viral proteins cannot explaiin the rapid onset of the adaptive immune response (Yewdell et al., 1996).

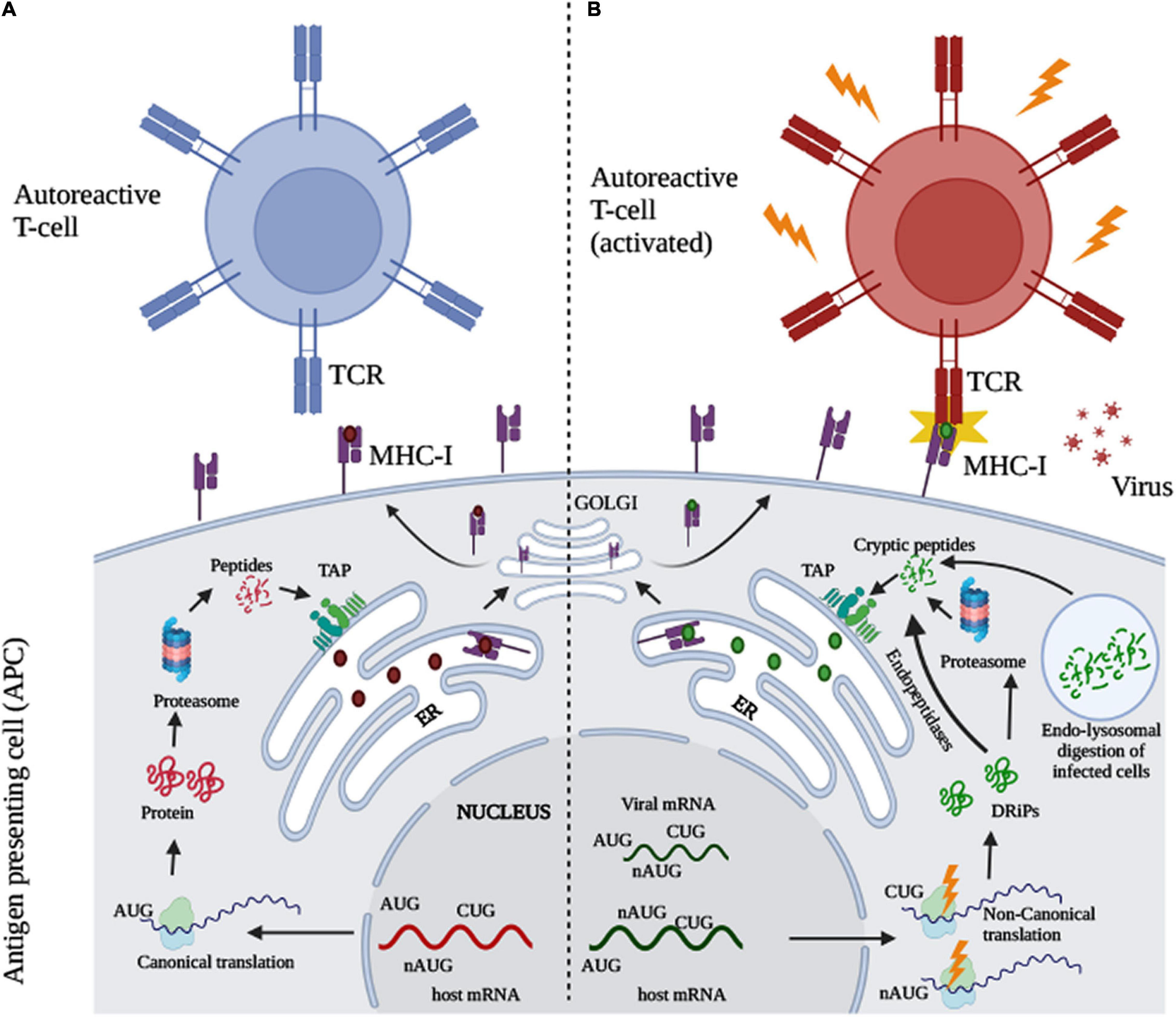
The following will be a gross simplification of fairly complex processes.
Your immune system does not possess the ability to recognize big chunks of protein, even parts of a pathogen are too big for your antigen-presenting cells (APC) to pick up, so they are often “digested” by certain immune cells, becoming smaller chucks, and at this point, your immune system can start working to recognize the pathogen and start doing its work.
During the normal function of the cell, following canonical (meaning tested, established, fairly well-understood mechanisms and pathways) your cell can “digest” these proteins, the peptides produced are loaded into the part of the cell that starts an immune response, but they fail to create an auto reaction. During viral infection, through many different pathways, creating a lot of stress inside and outside the cells and using noncanonical pathways, it creating novel peptides (previously hidden) that can now create a reaction with specific T-Cells, creating autoimmune responses.
The following quote from the second “Non-canonical Translation and Defective Ribosomal Products in Autoimmunity” explains it fairly well.
T-cell lymphocytes expressing either CD4+ or CD8+ TCRs are capable of recognizing antigen associated MHC class II and I molecules respectively (Kumar et al., 2018). During their development in the thymus, T-cells undergo positive and negative selection, where T-cells recognizing self-antigen MHC complexes with low to moderate affinity undergo proliferation, whereas strong interactions with high affinity self-antigen MHC complexes leads to cell death, preventing autoimmunity, while some of the cells differentiate into regulatory T-cells (Takaba and Takayanagi, 2017). The self-tolerant T-cells selected recognize abnormal or foreign peptides during infection and disease. However, some T-cells escape tolerance and are activated when exposed to self-peptides which may not have been expressed during their development at optimal levels. These “invisible” peptides are therefore cryptic in nature and may not be sufficiently expressed during T-cell selection (Sercarz et al., 1993; Lanzavecchia, 1995). In certain cases, cryptic epitopes would have lower affinity for MHC molecules and may not be presented due to competition with the high affinity epitopes. However, inflammatory stimuli and co-stimulatory signals might alter their expression and processing, thereby activating self-reactive T-cells.
The following is the reason I decided to introduce the concept and this paper now.
Indeed, evidence to define the role of cryptic peptides in virus-induced autoimmunity emerged from studies in HIV infections where the viral membrane protein, gp120 was able to modulate the processing of endogenous CD4+ to generate self-peptides (Salemi et al., 1995). Expression of gp120 led to an increase in CD4+ endocytic processing, generating novel epitopes which in turn generated anti-CD4 autoantibodies. Synthesis and presentation of less immunodominant and normally absent epitopes was also observed in the case of Theiler’s virus mediated MS (Miller et al., 1997). Differences in the processing and magnitude of antigen presentation between non-canonical and canonical peptides must play a crucial role. In the case of HSV-1-induced HSK, peptides originating from the viral protein, UL6 appeared to be cross-reactive when exposed to corneal T-cell clones in murine models (Zhao et al., 1998; Smatti et al., 2019). However, HSK studies in patients did not result in the isolation of T cells cross-reactive with UL6 indicating other mechanisms at play, which may involve self-antigens expressed at sufficiently high levels to trigger autoimmune T-cells (Verjans et al., 2000). Similarly, for EBV, anti-Ro antibodies found in SLE patients were cross-reactive with the EBV EBNA-1 protein (Poole et al., 2006). Synthetic peptides derived from Ro generated a broad range of antibodies against several epitopes including autoantibodies in mice, while immunization with intact Ro protein only produced antibodies against the immunizing peptide indicating differences in peptide processing during viral infections. While epitope spreading and mimicry explain the development of autoimmunity in viral infections, mechanistic differences in the generation of autoantigens must be addressed.
The most profound example for virus induced autoimmunity in the context of non-canonical translation comes from studies in IAV infection, where generation of viral DRiPs resulting from non-canonical translation through an ARF also results in the generation of a cellular ARF DRiP product (Zanker et al., 2019). The study described for the first time that viruses can induce cellular DRiPs, which has implications in understanding virus-induced autoimmunity. The authors conclude by stating that viruses might trigger autoimmunity by simply generating DRiPs. This might explain the preferential presentation of autoantigens under infection which generally remain hidden from developing T-cells. Perhaps a large amount of DRiPs derived peptides originate from non-canonical ORFs and remain invisible to the immune system under normal conditions that do not permit expression of such ORFs. Ribosome profiling conducted in human Beta cells in both normal and type 1 diabetes (T1D) conditions (Thomaidou et al., 2021) have revealed a novel set of non-canonical polypeptides which might unveil neo-antigens in T1D. Future studies to understand the role of novel ORFs in autoimmunity might explain several non-canonical autoantigens. Indeed, viruses encode many non-canonical ORFs that may play a role in better understanding virus-induced autoimmunity (Stern-Ginossar et al., 2012; Arias et al., 2014; Whisnant et al., 2020).
Influenza A Virus has the most profound, and quite a few numbers of novel mechanisms in regards to how it can affect how proteins are generated, and one such mechanism is by DRiPs (Defective Ribosomal Products, mentioned at the start), very small peptides that serve as “glue” (Ligands) for MHC I molecules, but recently uncovered novel mechanism in which can influence mRNA, miRNA, therefore affecting the proteins generated by cells. When generated via non-canonical mechanisms is the moment one starts rolling the autoimmunity dice.
Before I drive my final point, mostly leaving as a warning-slash-forecast, I leave the reader with the following.
Induction of broadly reactive influenza antibodies increases susceptibility to autoimmunity
Infection and vaccination repeatedly expose individuals to antigens that are conserved between influenza virus subtypes. Nevertheless, antibodies recognizing variable influenza epitopes greatly outnumber antibodies reactive against conserved epitopes. Elucidating factors contributing to the paucity of broadly reactive influenza antibodies remains a major obstacle for developing a universal influenza vaccine. Here, we report that inducing broadly reactive influenza antibodies increases autoreactive antibodies in humans and mice and exacerbates disease in four distinct models of autoimmune disease. Importantly, transferring broadly reactive influenza antibodies augments disease in the presence of inflammation or autoimmune susceptibility. Further, broadly reactive influenza antibodies spontaneously arise in mice with defects in B cell tolerance. Together, these data suggest that self-tolerance mechanisms limit the prevalence of broadly reactive influenza antibodies, which can exacerbate disease in the context of additional risk factors.

SARS-CoV-2 (the viral infection) to a minor degree may have caused B-cell defects in a small portion of the population, but most certainly it caused gradative, profound changes in the B Cell function of a considerable amount of people vaccinated with mRNA, to what degree and to actual end-effect, only time and (good) science will tell. Both virus and mRNA-Spike caused profound microbiome and immunological changes, shifting the body's immune response to a myriad of innocuous pathogens that now became rather troublesome for many people.
Given these changes, how the current iteration of mRNA functions, and all we know about it, and together with the information presented above, do you really think vaccinating against Influenza using mRNA is such a good idea ? I am not even talking about the probably trash-tier “protection” they will elicit but the complete autoimmune horror they most certainly will set off in due time. Many countries are now suspending the use of mRNA against Covid, going to the extent of stating even people at risk might not get a new booster in the next 12 months, but flu vaccines are always pushed hard, regardless of true efficacy.
Oh, I forgot this one. The Flu mRNA vaccine took incredible 8 DAYS to be designed.
Read the last paper I referenced again and remember, the biggest selling point for mRNA tech is the long-lasting high levels of antibodies. Sooner rather than later poorly thought out, badly designed rushed products will backfire, at so many non-linear levels it will be near impossible to piece everything together. To the benefit of the usual suspects.
I appreciate the support of those who choose a paid subscription, or who decide to buy me a coffee whenever they feel like it, and everyone who shares my Substack.
James was warning of this 3 years ago, pretty much.
Pathogenic priming likely contributes to serious and critical illness and mortality in COVID-19 via autoimmunity
James Lyons-Weiler
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7142689/
They know how to use chaos.
, "To the benefit of the usual suspects."